归一化
定义:将数据缩放到特定的区间中,以使得数值之间具有可比较性。
目标:
- 把数变为[0, 1]之间的数值
- 有量纲表达式变为无量纲表达式
为什么需要归一化:
- 提升模型的收敛速度。在梯度下降时,如果不同的特征的值范围差异很大,那么达到全局最小所需的步数就会很多。
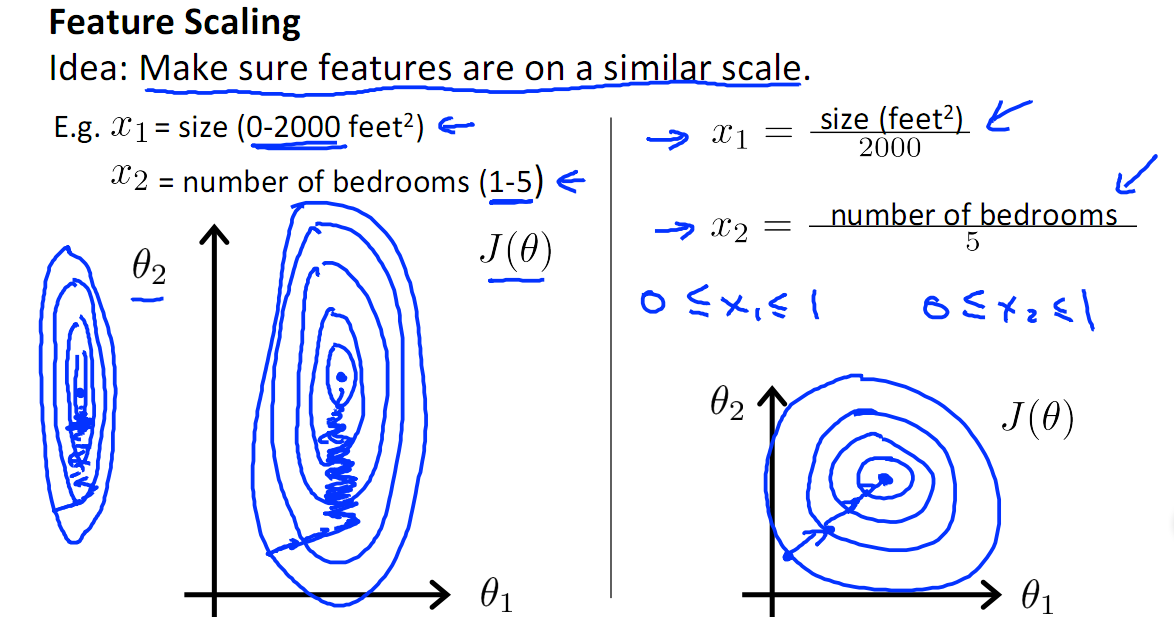
- 提升模型的精度。比如在计算相似性(距离等)时,不同数值范围的特征对距离值所产生的影响是不一样的(小的特征值可能不怎么影响),当进行归一化之后,能够使得不同的特征做出的贡献相同。
- 防止梯度爆炸。在神经网络中,梯度爆炸就是由于初始化权值过大,前面层会比后面层变化的更快,就会导致权值越来越大,梯度爆炸的现象就发生了。
哪些模型需要归一化:
- SVM:在特征缩放后,最优解与原来的不等价,所以如果不做缩放,模型参数会被较大或者较小的参数所主导。
- 逻辑回归:在特征缩放后,不会改变最优解,但是如果目标函数过于不均一,收敛会很慢,所以需要归一化。
- 神经网络、SGD:模型效果会强烈的依赖于归一化
哪些模型不需要归一化:
- 特征值在【0,1】之间的不再需要,否则会破坏其原始的稀疏性
- 决策树:效果不受归一化影响
- ICA:不需要归一化
- 最小二乘法OLS:不需要
方法
1. min-max normalization (Rescaling)
公式:\(X_{norm} = \frac{X - X_{min}}{X_{max}-X_{min}}\)
结果:将原始数据经过线性变换,把数值缩放到[0,1]之间,不会改变数据的分布。
2. Mean normalization
公式:\(X_{norm} = \frac{X - X_{mean}}{X_{max}-X_{min}}\)
结果:将原始数据经过线性变换,把数值缩放到[-1,1]之间
3. Standardization (标准化),z-score标准化,zero-mean normalization
公式:\(X_{norm} = \frac{X - X_{mean}}{\sigma}\),其中\(\sigma\)是标准差。
结果:转换后的数值,其平均值为0,方差为1,即服从标准正太分布,这个转换不会改变数据的分布。
4. Scaling to unit length
公式:\(X_{norm} = \frac{X}{\|X\|}\),其中\(\|X\|\)是这个数据向量的欧式长度(Euclidean length)。
结果:转换后的数值在[0,1]之间。
5. log函数转换
公式:\(X_{norm} = \frac{log{X}}{log{X_{max}}}\)
结果:转换后的数值在[0,1]之间。
6. atan函数转换
公式:\(X_{norm} = atan{X}*\frac{2}{\pi}\)
结果:如果X都大于0,则区间映射到[0,1],小于0的数据映射到[-1,0]之间。
7. quantile normalization
原理:
- 1)记录每个样本(列)中的每个数据的rank(原始rank);
- 2)每个样本,从小到达排序,计算排序后每个rank的平均值(排序后rank平均值);
- 3)根据原始rank从排序后rank平均值提取对应的值,取代原来的值,即为归一化之后的值。

结果:使得不同的数据集具有相同的分布,容易比较。(这个方法在microarray数据中使用得很多,原先叫quantile standardization,后来才叫做quantile normalization。wiki给出了一个例子,说的是在不同的样本中,如果将基因的表达量归一化到同一水平。)。下面是不同样本中前后表达量分布的例子: 
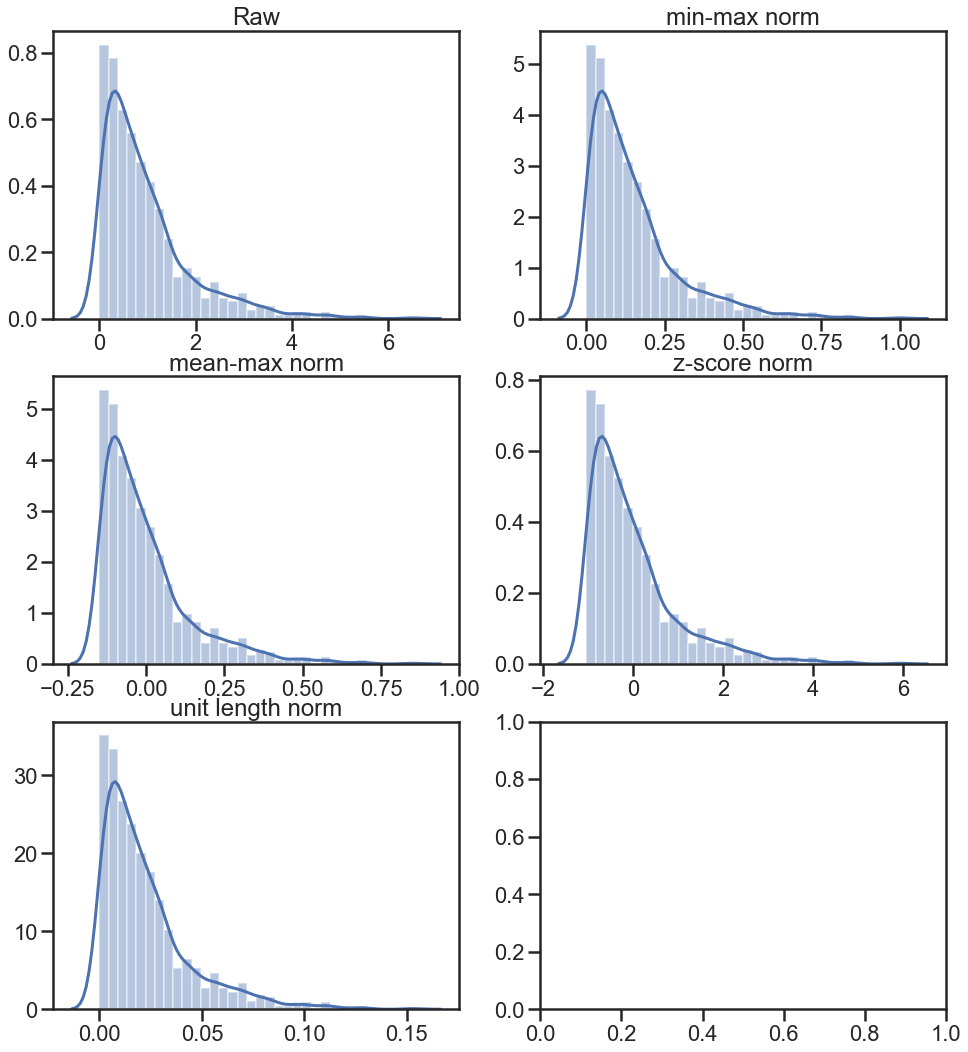
例子比较
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
sns.set_style('ticks')
np.random.random(42)
X = np.random.exponential(size=1000)
X_norm_min_max = (X - X.min()) / (X.max() - X.min())
X_norm_mean_max = (X - X.mean()) / (X.max() - X.min())
X_norm_zscore = (X - X.mean()) / X.std()
X_norm_unit = X / np.linalg.norm(X)
fig, ax = plt.subplots(3, 2, figsize=(16,18), sharex=False, sharey=False)
sns.distplot(X, ax=ax[0,0])
ax[0,0].set_title('Raw')
sns.distplot(X_norm_min_max, ax=ax[0,1])
ax[0,1].set_title('min-max norm')
sns.distplot(X_norm_mean_max, ax=ax[1,0])
ax[1,0].set_title('mean-max norm')
sns.distplot(X_norm_zscore, ax=ax[1,1])
ax[1,1].set_title('z-score norm')
sns.distplot(X_norm_unit, ax=ax[2,0])
ax[2,0].set_title('unit length norm')
参考
If you link this blog, please refer to this page, thanks!
Post link:https://tsinghua-gongjing.github.io/posts/normalization.html
Latest articles
Links
- ZhangLab , RISE database , THU life , THU info
- Data analysis: pandas , numpy , scipy
- ML/DL: sklearn , sklearn(中文) , pytorch
- Visualization: seaborn , matplotlib , gallery
- Github: me