概述
T 分布随机近邻嵌入(t-distributed Stochastic Neighbor Embedding):对高维的数据进行降维可视化,识别相关联的模式,保持局部结构(高维空间中距离相近的点投影到低维中扔然相近)。
原理
- 对数据点近邻分布进行建模。高维空间:高斯分布 =》低维空间(比如二维空间):t分布(更长的尾部,更均匀分布)。
- 下图红色为t分布,蓝色为正太分布:

- 目标:找到一个变换,将高维空间投射到二维空间,最小化所有点在这两个分布之间的差距。
- 困惑度(Perplexity):拟合分布时考虑的近邻数。低困惑度:最近的几个近邻。
- 分布时基于距离的,需要所有数据为数值型。类别变量:二值编码等进行转换。
- 随机近邻嵌入:
- 数据点的欧几里德距离转换为条件概率而表征相似性。如果两个点相近,则一个呗选为另一个的近邻的条件概率高。

- 引入矩阵(低维空间的)Y,同样计算条件概率:
.png)
- 整体目标是对于Y中的点,使其条件概率分布近似于原始高维空间,所以可以构建损失函数如下,并用梯度下降寻找最小值。
.png)
实现
因为tSNE在生物学的单细胞分析里比较多,所以利用已发表的数据重现一下文章的图。
- 文章:Jindal et al. Discovery of rare cells from voluminous single cell expression data. Nature Communications. 2018
- 数据:preprocessedData_jurkat_two_species_1580.txt.gz
sklearn版本
导入模块:
import seaborn as sns
import pandas as pd
import sklearn.manifold as skma
import gzip
import numpy as np
读取数据:
with gzip.GzipFile('./preprocessedData_jurkat_two_species_1580.txt.gz', 'r') as fid:
preprocessedData = np.genfromtxt(fid)
print type(preprocessedData)
print preprocessedData.shape
# <type 'numpy.ndarray'>
# (1580, 1000)
做tSNE并保存结果:
tsne = skma.TSNE()
projs = tsne.fit_transform(preprocessedData)
np.savetxt('preprocessedData_jurkat_two_species_1580.tsne.txt', projs)
可视化的结果:
f = './preprocessedData_jurkat_two_species_1580.tsne.txt'
df1 = pd.read_csv(f, header=None, sep=' ')
df1.columns = ['tSNE-1', 'tSNE-2']
df1.head()
sns.scatterplot(x='tSNE-1', y='tSNE-2', data=df1)
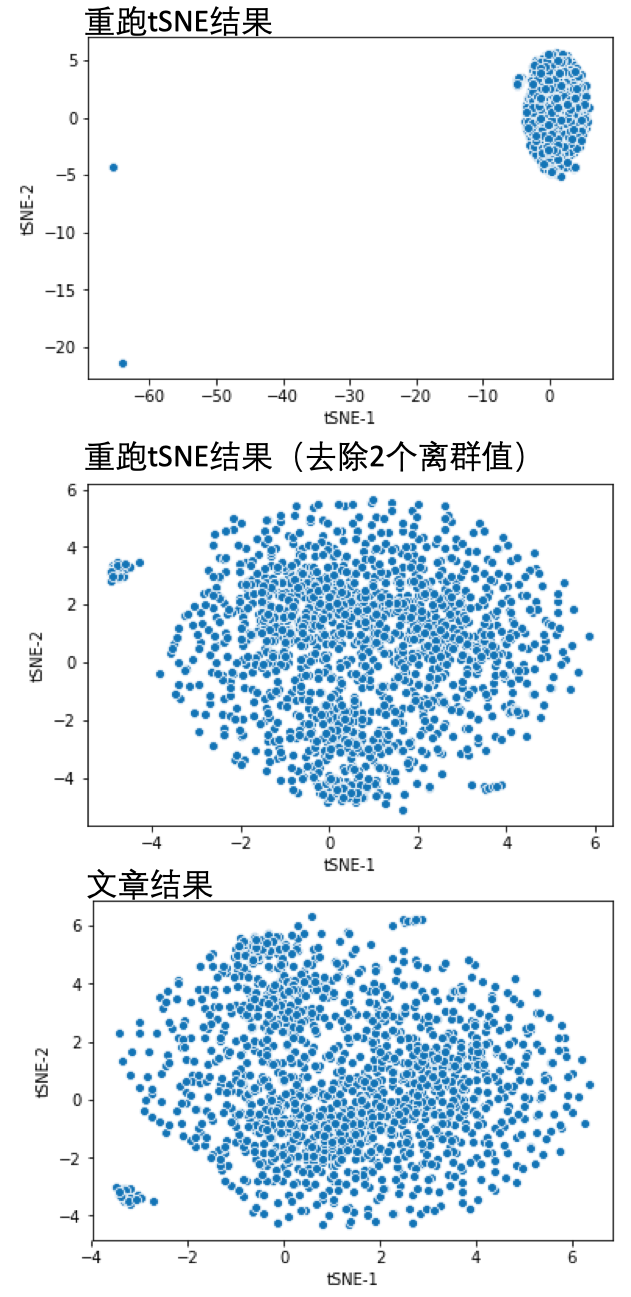
可以看到有两个离群值,将其去除后在可视化:
df1_filterOutliner = df1[df1['tSNE-1']>-20]
sns.scatterplot(x='tSNE-1', y='tSNE-2', data=df1_filterOutliner)
文章提供的结果:
f = './tsne_jurkat_1k_outlierRemoved.txt'
df2 = pd.read_csv(f, header=None, sep=' ')
df2.columns = ['tSNE-1', 'tSNE-2']
df2.head()
sns.scatterplot(x='tSNE-1', y='tSNE-2', data=df2)
可以看到,两个图不完全一样,因为tSNE函数会设置初始状态值,不同的值结果存在偏差,可以结合聚类的结果看是不是大致相似。
参考
If you link this blog, please refer to this page, thanks!
Post link:https://tsinghua-gongjing.github.io/posts/tSNE.html
Previous:
ICA独立成分分析
Latest articles
Links
- ZhangLab , RISE database , THU life , THU info
- Data analysis: pandas , numpy , scipy
- ML/DL: sklearn , sklearn(中文) , pytorch
- Visualization: seaborn , matplotlib , gallery
- Github: me