- Common tools for motif analysis
- FIMO error: base frequency
- Plot motif logo using meme2img
- TOMOM: identify motif against known motif source
- Prepare .meme file from .fa file
- Multiple peak region (.bed file) overlap
Common tools for motif analysis
-
Motif discovery:
- MEME: Multiple EM for Motif Elicitation, discovers novel, ungapped motifs (recurring, fixed-length patterns) in your sequences (sample output from sequences).
- DREME: Discriminative Regular Expression Motif Elicitation, discovers short, ungapped motifs (recurring, fixed-length patterns) that are relatively enriched in your sequences compared with shuffled sequences or your control sequences (sample output from sequences).
- MEME-ChIP: Motif Analysis of Large Nucleotide Datasets, performs comprehensive motif analysis (including motif discovery) on LARGE (50MB maximum) sets of sequences (typically nucleotide) such as those identified by ChIP-seq or CLIP-seq experiments.
-
Motif scanning:
- FIMO: Find Individual Motif Occurrences, scans a set of sequences for individual matches to each of the motifs you provide.
-
Motif comparison:
- TOMTOM: compares one or more motifs against a database of known motifs (e.g., JASPAR). Tomtom will rank the motifs in the database and produce an alignment for each significant match.
FIMO error: base frequency
根据指定的motif文件,对query的fasta文件,用fimo扫潜在的RBP binding site:
fimo -oc combine_all_full \
--thresh 0.001 \
/Share/home/zhangqf7/gongjing/zebrafish/script/zhangting/paris_RBP/motif_CISBP_RNA_narrow/Collapsed.meme \
combine_all.full.fa
motif .meme 文件的头文件(修改之前):
MEME version 4.10.1
ALPHABET= ACGU
Background letter frequencies (from uniform background):
A 0.25000 C 0.25000 G 0.25000 U 0.25000
报错:
Errors from MEME text parser:
The frequency letter U at position 4 is invalid for the DNA alphabet.
Errors from MEME XML parser:
Expected state IN_MEME not found!
MEME XML parser returned error code 4.
FATAL: No motifs could be read.
motif .meme 文件的头文件(修改之后):
MEME version 4.10.1
ALPHABET= ACGT
Background letter frequencies (from uniform background):
A 0.25000 C 0.25000 G 0.25000 T 0.25000
Plot motif logo using meme2img
[zhangqf7@loginview02 ]$ meme2images
Usage:
meme2images [options] <motifs file> <output directory>
Options:
-motif <ID> output only a selected motif; repeatable
-eps output logos in eps format
-png output logos in png format
-rc output reverse complement logos
-help print this usage message
Description:
Creates motif logos from any MEME or DREME motif format.
[zhangqf7@loginview02 MolCel2018_RBP]$ cat test.meme
MEME version 4.10.1
ALPHABET
P
Q
S
Z
END ALPHABET
strands: +
MOTIF DGCR8_1_str
letter-probability matrix: alength= 4 w= 8 nsites= 269 E= 3.3e-302
0.096655 0.111524 0.639405 0.152416
0.048326 0.022304 0.866174 0.063196
0.037174 0.044609 0.836433 0.081784
0.052045 0.066914 0.788104 0.092937
0.085501 0.078066 0.765801 0.070632
0.059479 0.130111 0.698886 0.111524
0.122676 0.122677 0.657993 0.096654
0.208179 0.193309 0.397770 0.200743
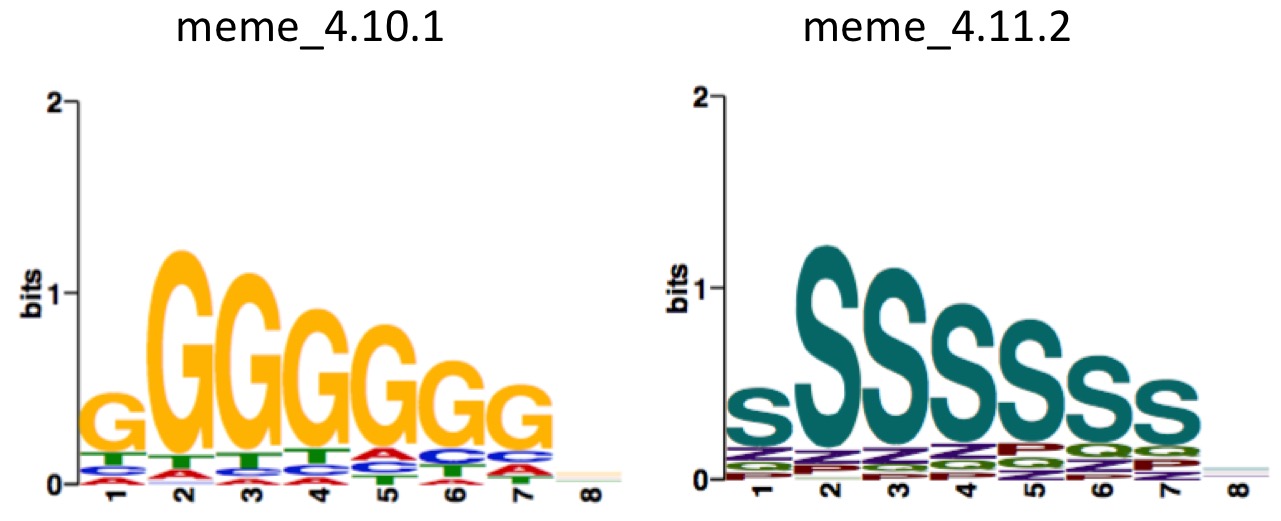
# generate wrong character
[zhangqf7@loginview02 MolCel2018_RBP]$ which meme2images
/Share/home/zhangqf/usr/meme_4.10.1/bin/meme2images
[zhangqf7@loginview02 MolCel2018_RBP]$ meme2images test.meme -eps test
# generate correct plot
[zhangqf7@loginview02 MolCel2018_RBP]$ /Share/home/zhangqf/usr/meme_4.11.2-app/bin/meme2images test.meme -eps test
TOMOM: identify motif against known motif source
[zhangqf7@loginview02 bin]$ /Share/home/zhangqf7/bin/tomtom --version
5.0.2
# 最新的版本比之前的版本添加了 -norc 参数
# 指定这个参数,只输出正链的,
# 对于扫RNA motif链的方向性很重要(一般不要负链上的)
[zhangqf7@loginview02 bin]$ /Share/home/zhangqf7/bin/tomtom
No query motif database supplied
Usage:
tomtom [options] <query file> <target file>+
Options:
-o <output dir> Name of directory for output files;
will not replace existing directory
-oc <output dir> Name of directory for output files;
will replace existing directory
-xalph Convert the alphabet of the target motif databases
to the alphabet of the query motif database
assuming the core symbols of the target motif
alphabet are a subset; default: reject differences
-bfile <background file>
Name of background file;
default: use the background from the query
motif database
-motif-pseudo <pseudo count>
Apply the pseudocount to the query and target motifs;
default: apply a pseudocount of 0.1
-m <id> Use only query motifs with a specified id;
may be repeated
-mi <index> Use only query motifs with a specifed index;
may be repeated
-thresh <float> Significance threshold; default: 0.5
-evalue Use E-value threshold; default: q-value
-dist allr|ed|kullback|pearson|sandelin|blic1|blic5|llr1|llr5
Distance metric for scoring alignments;
default: pearson
-internal Only allow internal alignments;
default: allow overhangs
-min-overlap <int>
Minimum overlap between query and target;
default: 1
-norc Do not score the reverse complements of targets
-incomplete-scores
Ignore unaligned columns in computing scores
default: use complete set of columns
-text Output in text format (default is HTML)
-png Create PNG logos; default: don't create PNG logos
-eps Create EPS logos; default: don't create EPS logos
-no-ssc Don't apply small-sample correction to logos;
default: use small-sample correction
-verbosity [1|2|3|4]
Set the verbosity of the program; default: 2 (normal)
-version Print the version and exit
$ tomtom -no-ssc
-oc ./tomom_5_Collapsed-used
-verbosity 1
-min-overlap 5
-dist pearson
-evalue
-thresh 5
./dreme.meme
/path/to/motif/source
Prepare .meme file from .fa file
Prepare the .fa file:
[zhangqf7@loginview02 motif_CISBP_RNA_narrow]$ head miR_Family_Info.fa
>let-7
GAGGUAG
>miR-1/206
GGAAUGU
>miR-10
ACCCUGU
>miR-101
ACAGUAC
>miR-103/107
GCAGCAU
The output .meme file (not in order as provides as using dict object):
[zhangqf7@loginview02 motif_CISBP_RNA_narrow]$ head -30 miR_Family_Info.meme
MEME version 4
ALPHABET= ACGT
Background letter frequencies (from uniform background):
A 0.25000 C 0.25000 G 0.25000 T 0.25000
MOTIF miR-214 (miR-214)
letter-probability matrix: alength= 4 w= 7 nsites= 20 E= 0
0 1 0 0
1 0 0 0
0 0 1 0
0 1 0 0
1 0 0 0
0 0 1 0
0 0 1 0
MOTIF miR-217 (miR-217)
letter-probability matrix: alength= 4 w= 7 nsites= 20 E= 0
1 0 0 0
0 1 0 0
0 0 0 1
0 0 1 0
0 1 0 0
1 0 0 0
0 0 0 1
MOTIF miR-740 (miR-740)
The script:
from pyfasta import Fasta
def read_fa(fa):
fa_dict1 = Fasta(fa, key_fn=lambda key: key.split("\t")[0])
fa_dict = {i.split()[0]: j[0:] for i, j in fa_dict1.items()}
print fa_dict.keys()[0:3]
return fa_dict
def write_pwm(seq=None, motif_name=None):
if seq is None:
seq = 'AGGUAAGU'
if motif_name is None:
motif_name = 'Novel'
s = 'MOTIF %s (%s)'%(motif_name, motif_name)
s += '\n\n'
s += 'letter-probability matrix: alength= 4 w= %s nsites= 20 E= 0'%(len(seq))
s += '\n'
for n in seq:
n_s = [1 if n.upper() == b else 0 for b in ['A', 'C', 'G', 'U']]
s += ' '+'\t'.join(map(str, n_s))
s += '\n'
s += '\n'
return s
def meme_header():
header = 'MEME version 4\n\nALPHABET= ACGT\n\nBackground letter frequencies (from uniform background):\nA 0.25000 C 0.25000 G 0.25000 T 0.25000\n\n'
return header
def write_fa_all(fa=None):
savefn = fa.replace('.fa', '.meme')
SAVEFN = open(savefn, 'w')
fa_dict = read_fa(fa)
s_all = ''
header = meme_header()
s_all += header
for i in fa_dict:
seq = fa_dict[i]
s = write_pwm(seq, motif_name=i)
s_all += s
print >>SAVEFN, s_all
SAVEFN.close()
def main():
# header = meme_header()
# s = write_pwm()
# print header + s
write_fa_all()
if __name__ == '__main__':
main()
Multiple peak region (.bed file) overlap
-
biostar: Tutorial: Visualization of ChIP-Seq peak overlaps using HOMER mergePeaks and UpSetR
-
github: Calling mergePeaks to calculate overlap and UpSetR to plot
-
HOMER mergePeaks: Finding Overlapping and Differentially Bound Peaks
# Quick example
# calculate overlapping
mergePeaks H3K27AC.bed \
H3K27ME3.bed \
H3K4ME3.bed \
H3K9AC.bed \
gencode.bed \
-prefix mergepeaks \
-venn venn.txt \
-matrix matrix.txt
# plot overlapping
# SampleID:: output file name (same dir as venn.txt)
Rscript --vanilla multi_peaks_UpSet_plot.R "SampleID" venn.txt
The number of merged peaks are less than original peaks number, as overlapping regions will bed merged (asked in biostar: HOMER mergePeaks totals don’t add up to peaks input):
# original: two entry region overlapped
NM_001017557 1092 1121 NM_001017557 84 1373 + cds *
NM_001017557 1111 1139 NM_001017557 84 1373 + cds *
# merge: the two regions are merged
# in this case, the final number of entry is <= original
Merged-NM_001017557-1116-2 NM_001017557 1093 1139 + 0.000000 window.bed 2 NM_001017557--18,NM_001017557--19
The example venn.txt file output is as below:
H3K27AC.bed H3K27ME3.bed H3K4ME3.bed H3K9AC.bed gencode.bed Total Name
X X 1690 H3K27AC.bed|H3K27ME3.bed
X X X 758 H3K27AC.bed|H3K27ME3.bed|gencode.bed
X X X 147 H3K27AC.bed|H3K27ME3.bed|H3K9AC.bed
X X X X 87 H3K27AC.bed|H3K27ME3.bed|H3K9AC.bed|gencode.bed
X X X 1420 H3K27AC.bed|H3K27ME3.bed|H3K4ME3.bed
X X X X 1797 H3K27AC.bed|H3K27ME3.bed|H3K4ME3.bed|gencode.bed
X X X X 1187 H3K27AC.bed|H3K27ME3.bed|H3K4ME3.bed|H3K9AC.bed
We can also calculate the cross-way percentage using these scripts, which is comparable with the section Display cross-way percentage for each set
import pandas as pd
from nested_dict import nested_dict
import matplotlib as mpl
mpl.use('Agg')
import matplotlib.pyplot as plt
import seaborn as sns
import matplotlib.colors as mcolors
sns.set(style="ticks")
sns.set_context("poster")
def load_upset_file(txt=None, header_ls=None,):
if txt is None:
txt = '/Share/home/zhangqf7/gongjing/zebrafish/result/dynamic_merge_region/005_005_new/abs/venn.txt'
set_category_stat_dict = nested_dict(2, int)
df = pd.read_csv(txt, header=0, sep='\t')
for sample in df.columns[0:-2]:
for label,count,name in zip(df[sample], df['Total'], df['Name']):
if label == 'X' and sample in name:
set_category_stat_dict[sample][len(name.split('|'))] += count
set_category_stat_df = pd.DataFrame.from_dict(set_category_stat_dict, orient='index')
set_category_stat_df = set_category_stat_df.loc[df.columns[0:-2], :]
print set_category_stat_df
set_category_stat_df_ratio = set_category_stat_df.div(set_category_stat_df.sum(axis=1), axis=0)
print set_category_stat_df_ratio
fig, ax = plt.subplots(1, 2)
set_category_stat_df.plot(kind='bar', stacked=True, ax=ax[0])
set_category_stat_df_ratio.plot(kind='bar', stacked=True, ax=ax[1])
plt.tight_layout()
savefn = txt.replace('.txt', '.ratio.pdf')
plt.savefig(savefn)
plt.close()
def main():
load_upset_file()
if __name__ == '__main__':
main()
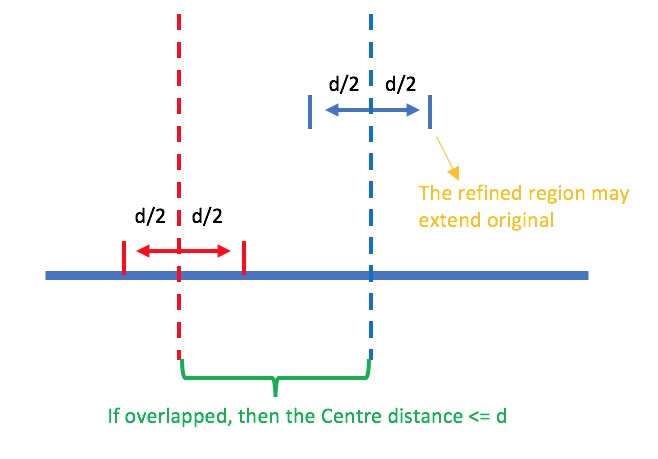
What’s the meaning of -d parameter:
HOMER manual: Maximum distance between peak centers to merge, default: 100
Also asked in biostar HOMER mergePeaks -d value definition: no answer till now.
Prepare the test region bed file:
[zhangqf7@bnode02 abs]$ cat test1.bed
ch1 100 150
[zhangqf7@bnode02 abs]$ cat test2.bed
ch1 70 90
Run using -d 10, the two region are not merged:
[zhangqf7@bnode02 abs]$ mergePeaks test1.bed test2.bed -prefix test10 -venn test10.venn.txt -d 10
Max distance to merge: 10 bp
Merging peaks...
Comparing test1.bed (1 total) and test1.bed (1 total)
Comparing test1.bed (1 total) and test2.bed (1 total)
Comparing test2.bed (1 total) and test1.bed (1 total)
Comparing test2.bed (1 total) and test2.bed (1 total)
[zhangqf7@bnode02 abs]$ cat test10_test1.bed
#name (cmd = mergePeaks test1.bed test2.bed -prefix test10 -venn test10.venn.txt -d 10) chr start end strand Stat Parent files Total subpeakstest1.bed test2.bed
Merged-ch1-125-1 ch1 120 130 + 0.000000 test1.bed 1 default-1
[zhangqf7@bnode02 abs]$ cat test10_test2.bed
#name (cmd = mergePeaks test1.bed test2.bed -prefix test10 -venn test10.venn.txt -d 10) chr start end strand Stat Parent files Total subpeakstest1.bed test2.bed
Merged-ch1-80-1 ch1 75 85 + 0.000000 test2.bed 1 default-1
[zhangqf7@bnode02 abs]$ cat test10.venn.txt
test1.bed test2.bed Total Name
X 1 test2.bed
X 1 test1.bed
Run using -d 35, the two region are still not merged:
[zhangqf7@bnode02 abs]$ mergePeaks test1.bed test2.bed -prefix test35 -venn test35.venn.txt -d 35
Max distance to merge: 35 bp
Merging peaks...
Comparing test1.bed (1 total) and test1.bed (1 total)
Comparing test1.bed (1 total) and test2.bed (1 total)
Comparing test2.bed (1 total) and test1.bed (1 total)
Comparing test2.bed (1 total) and test2.bed (1 total)
[zhangqf7@bnode02 abs]$ cat test35.venn.txt
test1.bed test2.bed Total Name
X 1 test2.bed
X 1 test1.bed
For -d less equal than 45, the regions are not merged.
[zhangqf7@bnode02 abs]$ ll test45*
-rw-rw----+ 1 zhangqf7 zhangqf 225 Sep 28 20:00 test45_test1.bed
-rw-rw----+ 1 zhangqf7 zhangqf 223 Sep 28 20:00 test45_test2.bed
-rw-rw----+ 1 zhangqf7 zhangqf 61 Sep 28 20:00 test45.venn.txt
[zhangqf7@bnode02 abs]$ cat test45_test1.bed
#name (cmd = mergePeaks test1.bed test2.bed -prefix test45 -venn test45.venn.txt -d 45) chr start end strand Stat Parent files Total subpeaks test1.bed test2.bed
Merged-ch1-125-1 ch1 103 147 + 0.000000 test1.bed 1 default-1
[zhangqf7@bnode02 abs]$ cat test45_test2.bed
#name (cmd = mergePeaks test1.bed test2.bed -prefix test45 -venn test45.venn.txt -d 45) chr start end strand Stat Parent files Total subpeaks test1.bed test2.bed
Merged-ch1-80-1 ch1 58 102 + 0.000000 test2.bed 1 default-1
If we set -d >=46 here, here is the merged result:
[zhangqf7@bnode02 abs]$ mergePeaks test1.bed test2.bed -prefix test46 -venn test46.venn.txt -d 46
Max distance to merge: 46 bp
Merging peaks...
Comparing test1.bed (1 total) and test1.bed (1 total)
Comparing test1.bed (1 total) and test2.bed (1 total)
Comparing test2.bed (1 total) and test1.bed (1 total)
Comparing test2.bed (1 total) and test2.bed (1 total)
[zhangqf7@bnode02 abs]$ ll test46*
-rw-rw----+ 1 zhangqf7 zhangqf 243 Sep 28 20:01 test46_test1.bed_test2.bed
-rw-rw----+ 1 zhangqf7 zhangqf 57 Sep 28 20:01 test46.venn.txt
[zhangqf7@bnode02 abs]$ cat test46_test1.bed_test2.bed
#name (cmd = mergePeaks test1.bed test2.bed -prefix test46 -venn test46.venn.txt -d 46) chr start end strand Stat Parent files Total subpeaks test1.bed test2.bed
Merged-ch1-102-2 ch1 57 148 + 0.000000 test1.bed|test2.bed 2 default-1 default-1
[zhangqf7@bnode02 abs]$ cat test46.venn.txt
test1.bed test2.bed Total Name
X X 1 test1.bed|test2.bed
看起来是这样的,当设置一个-d时,会对每一个peak用一个长度等于d的region去替代,然后看替代后的region之间是否有overlap(不是这个的centre),如果有overlap,就merge在一起。因为在-d=45时,替代后的region,首尾相差很小,但是没有overlap,所以没有merge,而取更大的时候,就merge在一起了。如其manual所写,peak centre的距离小于d时会被合并,合并后的起始很大程度会发生改变。
If you link this blog, please refer to this page, thanks!
Post link:https://tsinghua-gongjing.github.io/posts/MEME-suit.html
Latest articles
Links
- ZhangLab , RISE database , THU life , THU info
- Data analysis: pandas , numpy , scipy
- ML/DL: sklearn , sklearn(中文) , pytorch
- Visualization: seaborn , matplotlib , gallery
- Github: me