Linux common used tricks
2018-06-19
- Rename multiple filenames reference
- Drop specific columns linuxconfig
- Mac OS general compress/decompress command line tool
- Iterate over two arrays stackoverflow
- GCC version
- Print every two lines as one line
- Update file soft link
- install software instead of default dir
- 脚本中获得当前路径及文件夹名称
- 获取脚本参数
- ImportError: /lib64/libc.so.6: version `GLIBC_2.14’ not found
ssh免密码登录- rsync同步文件
- linux更改终端颜色
- 常用alias
Rename multiple filenames reference
# rename 's/old-name/new-name/' files
gongjing@hekekedeiMac ..ject/meme_img % ll
total 1.3M
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 logoRBNS_A1CF.eps
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 logoRBNS_BOLL.eps
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 logoRBNS_CELF1.eps
gongjing@hekekedeiMac ..ject/meme_img % rename 's/logoRBNS_//' *eps
gongjing@hekekedeiMac ..ject/meme_img % ll
total 1.3M
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 A1CF.eps
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 BOLL.eps
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 CELF1.eps
gongjing@hekekedeiMac ..ject/meme_img % rename 's/.eps//' *eps
gongjing@hekekedeiMac ..ject/meme_img % ll
total 1.3M
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 A1CF
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 BOLL
-rw-r--r-- 1 gongjing staff 14K Jun 19 14:47 CELF1
有的时候rename命令不正常,可能是因为使用的命令来源不同,就像这里讨论的:
首先要查看环境的rename是来源,然后写对应的命令,不能弄错了:
$ rename --version
/usr/bin/rename using File::Rename version 0.20
$ rename 's/\.jpeg$/.jpg/' *
$ rename --version
rename from util-linux 2.30.2
$ rename .jpeg .jpg *
# 集群上使用的是util-linux这个版本
# 命令:rename pattern replace files
$ rename --version
rename (util-linux-ng 2.17.2)
Drop specific columns linuxconfig
### use cut -f and --complement
[zhangqf5@loginview02 RBPgroup_CLIPseq_hg38_anno]$ head test.txt|awk -F "\t" '{print NF}'
31
31
31
31
31
31
31
31
31
31
[zhangqf5@loginview02 RBPgroup_CLIPseq_hg38_anno]$ head test.txt|awk 'BEGIN{OFS="\t";}{if($17==10)print}'|cut -f17 --complement|awk -F "\t" '{print NF}'
30
30
30
30
30
30
30
30
Mac OS general compress/decompress command line tool
# https://theunarchiver.com/command-line
$ wget https://cdn.theunarchiver.com/downloads/unarMac.zip
$ ll
-rwxr-xr-x 1 gongjing staff 1.8M May 19 2016 lsar
-rwxr-xr-x 1 gongjing staff 1.8M May 19 2016 unar
$ unar
unar v1.10.1 (May 19 2016), a tool for extracting the contents of archive files.
Usage: unar [options] archive [files ...]
Available options:
-output-directory (-o) <string> The directory to write the contents of the archive to. Defaults to the current directory. If set to a single dash (-), no files will be
created, and all data will be output to stdout.
-force-overwrite (-f) Always overwrite files when a file to be unpacked already exists on disk. By default, the program asks the user if possible, otherwise skips
the file.
-force-rename (-r) Always rename files when a file to be unpacked already exists on disk.
-force-skip (-s) Always skip files when a file to be unpacked already exists on disk.
-force-directory (-d) Always create a containing directory for the contents of the unpacked archive. By default, a directory is created if there is more than one
top-level file or folder.
-no-directory (-D) Never create a containing directory for the contents of the unpacked archive.
-password (-p) <string> The password to use for decrypting protected archives.
-encoding (-e) <encoding name> The encoding to use for filenames in the archive, when it is not known. If not specified, the program attempts to auto-detect the encoding
used. Use "help" or "list" as the argument to give a listing of all supported encodings.
-password-encoding (-E) <name> The encoding to use for the password for the archive, when it is not known. If not specified, then either the encoding given by the -encoding
option or the auto-detected encoding is used.
-indexes (-i) Instead of specifying the files to unpack as filenames or wildcard patterns, specify them as indexes, as output by lsar.
-no-recursion (-nr) Do not attempt to extract archives contained in other archives. For instance, when unpacking a .tar.gz file, only unpack the .gz file and not
its contents.
-copy-time (-t) Copy the file modification time from the archive file to the containing directory, if one is created.
-no-quarantine (-nq) Do not copy Finder quarantine metadata from the archive to the extracted files.
-forks (-k) <fork|visible|hidden|skip> How to handle Mac OS resource forks. "fork" creates regular resource forks, "visible" creates AppleDouble files with the extension ".rsrc",
"hidden" creates AppleDouble files with the prefix "._", and "skip" discards all resource forks. Defaults to "fork".
-quiet (-q) Run in quiet mode.
-version (-v) Print version and exit.
-help (-h) Display this information.
Iterate over two arrays stackoverflow
#!/bin/bash
array=( "Vietnam" "Germany" "Argentina" )
array2=( "Asia" "Europe" "America" )
for ((i=0;i<${#array[@]};++i)); do
printf "%s is in %s\n" "${array[i]}" "${array2[i]}"
done
GCC version
Run binary command which depends on GLIBC_2.14 and GLIBC_2.17:
[zhangqf5@loginview02 bin]$ ./clan_annotate
./clan_annotate: /lib64/libc.so.6: version `GLIBC_2.14' not found (required by ./clan_annotate)
./clan_annotate: /lib64/libc.so.6: version `GLIBC_2.17' not found (required by ./clan_annotate)
Check GCC version:
[zhangqf5@loginview02 bin]$ ldd --version
ldd (GNU libc) 2.12
Copyright (C) 2010 Free Software Foundation, Inc.
This is free software; see the source for copying conditions. There is NO
warranty; not even for MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE.
Written by Roland McGrath and Ulrich Drepper.
Print every two lines as one line
# print lines of files matched with specific pattern
[zhangqf7@loginview02 bwa]$ wl /Share/home/zhangqf7/jinsong_zhang/zebrafish/data/iclip/20181224/Rawdata/shi-zi-*/bwa/CTK_Procedure{1,2,3,4}/CITS/iCLIP.tag*p05.bed|awk '{print $1}'
15450
11876
26860
19994
69867
174233
13396
10204
24288
18042
65161
161563
2059
1369
1983
1569
6291
13434
2329
1535
2171
1758
6762
14614
666808
[zhangqf7@loginview02 bwa]$ wl /Share/home/zhangqf7/jinsong_zhang/zebrafish/data/iclip/20181224/Rawdata/shi-zi-*/bwa/CTK_Procedure{1,2,3,4}/CITS/iCLIP.tag*p05.bed|awk '{print $1}'|xargs -n4 -d'\n'
15450 11876 26860 19994
69867 174233 13396 10204
24288 18042 65161 161563
2059 1369 1983 1569
6291 13434 2329 1535
2171 1758 6762 14614
666808
Update file soft link
Use option -sfn of ln command as discussed here:
ln -sfn {path/to/file-name} {link-name}
install software instead of default dir
./configure --prefix=/somewhere/else/than/usr/local
make
make install
脚本中获得当前路径及文件夹名称
# get full path
work_space=$(pwd)
# get dir name instead of full path
work_dir_name=${PWD##*/}
获取脚本参数
参考这里:
# 这里第一个参数是GPU ID,第2个及之后的参数是要训练的文件夹名称(遍历训练)
for i in "${@:2}"
do
echo "process: "$i", with GPU: "$1
cd $expr/$i
bash train.sh $1
done
# $1: 第一个参数
# "${@:2}": 第二个及之后的所有参数
# "$@": 获得所有参数
# "${@:3:4}": 第3个参数开始的4个参数,即$3,$4,$5,$6
ImportError: /lib64/libc.so.6: version `GLIBC_2.14’ not found
### https://zhuanlan.zhihu.com/p/40444240
# 检查系统含有的GLIBC版本
$ strings /lib64/libc.so.6 |grep GLIBC
GLIBC_2.2.5
GLIBC_2.2.6
GLIBC_2.3
GLIBC_2.3.2
GLIBC_2.3.3
GLIBC_2.3.4
GLIBC_2.4
GLIBC_2.5
GLIBC_2.6
GLIBC_2.7
GLIBC_2.8
GLIBC_2.9
GLIBC_2.10
GLIBC_2.11
GLIBC_2.12
GLIBC_PRIVATE
# 下载、安装
$ wget https://ftp.gnu.org/gnu/glibc/glibc-2.14.tar.gz
$ tar zxf glibc-2.14.tar.gz
$ cd glibc-2.14
$ mkdir build
$ cd build
$ ../configure --prefix=/opt/glibc-2.14
$ make -j4
$ make install
# 添加到LD_LIBRARYPATH
$ export LD_LIBRARY_PATH=/opt/glibc-2.14/lib:$LD_LIBRARY_PATH
ssh免密码登录
网上教程很多,随便参考,比如这里:
在自己的Linux系统上生成SSH密钥和公钥
➜ ~ ssh-keygen
Generating public/private rsa key pair.
Enter file in which to save the key (/home/gongjing/.ssh/id_rsa):
/home/gongjing/.ssh/id_rsa already exists.
Overwrite (y/n)? y
Enter passphrase (empty for no passphrase):
Enter same passphrase again:
Your identification has been saved in /home/gongjing/.ssh/id_rsa.
Your public key has been saved in /home/gongjing/.ssh/id_rsa.pub.
The key fingerprint is:
SHA256:RWPGrhUHIbbp9DvzJX2cUOrzyFyibEdd8qU/7Mi5KUU gongjing@omnisky
The key's randomart image is:
+---[RSA 2048]----+
| o.Bo |
| . Bo.. |
| +..o . |
| o oo E+ o|
| So. .o.+o|
| . ..+ooo|
| + ++++o|
| .*=+X+.|
| .o+Xoo.|
+----[SHA256]-----+
将SSH公钥上传到Linux服务器(写在对方的~/.ssh/authorized_keys中)
➜ ~ ssh-copy-id gongjing@10.10.91.12
/usr/bin/ssh-copy-id: INFO: Source of key(s) to be installed: "/home/gongjing/.ssh/id_rsa.pub"
/usr/bin/ssh-copy-id: INFO: attempting to log in with the new key(s), to filter out any that are already installed
/usr/bin/ssh-copy-id: INFO: 1 key(s) remain to be installed -- if you are prompted now it is to install the new keys
gongjing@10.10.91.12's password:
Number of key(s) added: 1
Now try logging into the machine, with: "ssh 'gongjing@10.10.91.12'"
and check to make sure that only the key(s) you wanted were added.
如果id_rsa之前是其他用户(比如root)创建的,有可能会出现权限错误:
➜ ~ ssh gongjing@10.10.91.12
@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@
@ WARNING: UNPROTECTED PRIVATE KEY FILE! @
@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@
Permissions 0755 for '/home/gongjing/.ssh/id_rsa' are too open.
It is required that your private key files are NOT accessible by others.
This private key will be ignored.
Load key "/home/gongjing/.ssh/id_rsa": bad permissions
gongjing@10.10.91.12's password:
此时可以更改权限,使得文件id_rsa只有自己可读写(参考这里):
chmod 600 ~/.ssh/id_rsa
rsync同步文件
使用方法可参考这里:
- 参数
-a:迭代同步,子目录、文件链接等都会同步 - 参数
-P:显示同步进度,速率、已完成、剩余等
# 同步文件夹
rsync -aP download_20191204 gongjing@10.10.91.12:/home/gongjing/
# 同步文件
rsync -aP ./Untitled.ipynb gongjing@10.10.91.12:/home/gongjing/scripts
linux更改终端颜色
参考这里:
if [ "$color_prompt" = yes ]; then
PS1='${debian_chroot:+($debian_chroot)}\[\033[01;32m\]\u@\h\[\033[00m\]:\[\033[01;34m\]\w\[\033[00m\]\$ '
else
PS1='${debian_chroot:+($debian_chroot)}\u@\h:\w\$ '
fi
unset color_prompt force_color_prompt
PS1='${debian_chroot:+($debian_chroot)}\[\033[01;33;1m\]\u@\h\[\033[00m\]:\[\033[01;34m\]\w\[\033[00m\] \$ '
常用alias
# User specific aliases and functions
alias vb='vi ~/.bashrc'
alias sb='source ~/.bashrc'
alias les='less -S'
alias wl='wc -l'
alias lt='ls -lht'
alias ll='ls -lh'
Read full-text »
Think Stats: descriptive statistics?
2018-06-10
描述性统计量:
- 均值(mean):值的总和除以值的数量;平均值(average):若干种可以用于描述样本的典型值或集中趋势(central tendency)的汇总统计量之一。注意根据样本的范围选择合适的描述量。
- 方差:描述分散情况。方差(variance)的计算公式:\(\begin{align}\sigma^2 = \frac{1}{n}\sum_i(X_i-\mu)^2\end{align}\),其中\(\begin{align}X_i-\mu\end{align}\)叫做离均差(deviation from the mean),方差的平方根σ叫做标准差(Standard Deviation)。
- 分布(distribution):数据值出现的频繁程度。展示方法:直方图(histogram),展示频数或者概率(频数除以总数,归一化normalization的过程)。归一化之后的直方图称为PMF(Probability Mass Function,概率质量函数):值到概率的映射。
- 绘制PMF:直方图:利于展示众数、形状、异常值,数目较少时;折现图:数目较多。数目是指取值。
- 均值和方差也可以通过概率质量函数计算获得。均值:\(\begin{align} \mu = \sum_ip_ix_i \end{align}\),方差:\(\begin{align} \sigma^2 = \sum_ip_i(x_i-\mu)^2 \end{align}\)
- 异常值(outliner):数据采集或处理的错误,罕见结果,需要去除掉(trim)。
- 相对风险(relative risk):代表两个概率的比值。
- 条件概率(conditional probability):依赖于某个条件的概率。限定了某些条件(已知了这些),对应事件的概率会发生改变。
- 汇报结果:如何汇报结果还取决于具体目的。如果是要证明某种影响的显著性,可以选择汇总统计量,如强调差异的相对风险。如果是要说服某个患者,则可以选择能反映特定情况下差异的统计量。
术语:
- 区间(bin) 将相近数值进行分组的范围。
- 集中趋势(central tendency) 样本或总体的一种特征,直观来说就是最能代表平均水平的值。
- 临床上有重要意义(clinically significant) 分组间差异等跟实践操作有关的结果。
- 条件概率(conditional probability) 某些条件成立的情况下计算出的概率。
- 分布(distribution) 对样本中的各个值及其频数或概率的总结。
- 频数(frequency) 样本中某个值的出现次数。
- 直方图(histogram) 从值到频数的映射,或者表示这种映射关系的图形。
- 众数(mode) 样本中频数最高的值。
- 归一化(normalization) 将频数除以样本大小得到概率的过程。
- 异常值(outlier) 远离集中趋势的值。
- 概率(probability) 频数除以样本大小即得到概率。
- 概率质量函数(Probability Mass Function,PMF) 以函数的形式表示分布,该函数将值映射到概率。
- 相对风险(relative risk) 两个概率的比值,通常用于衡量两个分布的差异。
- 分散(spread) 样本或总体的特征,直观来说就是数据的变动有多大。
- 标准差(standard deviation) 方差的平方根,也是分散的一种度量。
- 修剪(trim) 删除数据集中的异常值。
- 方差(variance) 用于量化分散程度的汇总统计量。
Read full-text »
Think Stats: what is statistical thinking?
2018-06-04
Here is the note of reading Think Stats. The Chinese version online is here
统计思维
- 概率论:研究随机事件
- 统计学:根据数据样本推测总体情况
- 计算:量化分析
- 经验之谈(anecdotal evidence):基于个人感受的非公开数据(比如第一个孩子在预产期前出生)。缺点:1)观察的数量太少;2)选择偏差;3)确认偏差;4)不准确。
- 统计方法:基本步骤,1)收集数据;2)描述性统计;3)探索性数据分析:寻找模式、差异等;4)假设检验:影响的真实性;5)估计。
- 横断面研究(cross-sectional study):一群人在某个时间点的情况;纵贯研究(longitudinal study):在一段时间内反复观察同一群人。采用的数据集全国家庭成长调查(NSFG)属于前者。进行次数:周期(cycle);参与调查的人称为被调查者(respondent),一组被调查者就称为队列(cohort)。
- 被调查者文件中的每一行都表示一个被调查者。这行信息称为一条记录(record),组成记录的变量称为字段(field),若干记录的集合就组成了一个表(table)。
- 观察到的差异称为直观效应(apparent effect),不确定是都一定有意思的事情发生了。
术语:
- 经验之谈(anecdotal evidence) 个人随意收集的证据,而不是通过精心设计并经过研究得到的。
- 直观效应(apparent effect) 表示发生了某种有意思的事情的度量或汇总统计量。
- 人为(artifact) 由于偏差、测量错误或其他错误导致的直观效应。
- 队列(cohort) 一组被调查者。
- 横断面研究(cross-sectional study) 收集群体在特定时间点的数据的研究。
- 字段(field) 数据库中组成记录的变量名称。
- 纵贯研究(longitudinal study) 跟踪群体,随着时间推移对同一组人反复采集数据的研究。
- 过采样(oversampling) 为了避免样本量过少,而增加某个子群体代表的数量。
- 总体(population) 要研究的一组事物,通常是一群人,但这个术语也可用于动物、蔬菜和矿产。
- 原始数据(raw data) 未经或只经过很少的检查、计算或解读而采集和重编码的值。
- 重编码(recode) 通过对原始数据进行计算或是其他逻辑处理得到的值。
- 记录(record) 数据库中关于一个人或其他对象的信息的集合。
- 代表性(representative) 如果人群中的每个成员都有同等的机会进入样本,那么这个样本就具有代表性。
- 被调查者(respondent) 参与调查的人。
- 样本(sample) 总体的一个子集,用于收集数据。
- 统计显著(statistically significant) 若一个直观效应不太可能是由随机因素引起的,就是统计显著的。
- 汇总统计量(summary statistic) 通过计算将一个数据集归结到一个数字(或者是少量的几个数字),而这个数字能表示数据的某些特点。
- 表(table) 数据库中若干记录的集合。
Read full-text »
Comparison between Python and R
2018-05-30
NumPy for R (and S-Plus) users
This resource compare the basic usage in both languages, which is pretty direct.
R usage
字符串替换
函数:gsub(pattern, replacement, x),例子如下:
group <- c("12357e", "12575e", "197e18", "e18947")
group
[1] "12357e" "12575e" "197e18" "e18947"
gsub("e", "", group)
[1] "12357" "12575" "19718" "18947"
R脚本获取命令行参数
函数:commandArgs,例子如下:
args <- commandArgs(trailingOnly = TRUE)
print(args[1]) # args[0] -> script anme
判断字符串收尾是否含有特定字符
函数:startsWith,endsWith,例子如下:
> startsWith("what", "wha")
[1] TRUE
> startsWith("what", "ha")
[1] FALSE
读取文件时列名称保持原有特殊字符
函数:read.table的参数check.names默认对于特殊字符是要进行转换的,保持的例子如下:
read.csv(file, sep=",", header=T, check.names = FALSE)
示例脚本:含有spike-ins的RNAseq差异分析
args<-commandArgs(TRUE)
library(RUVSeq)
# f='Control_MO_H4_H6.txt'
f=args[1]
zfGenes = read.table(f, header=T, row.names=1, sep='\t')
filter <- apply(zfGenes, 1, function(x) length(x[x>5])>=2)
filtered <- zfGenes[filter,]
genes <- rownames(filtered)[grep("^N", rownames(filtered))]
spikes <- rownames(filtered)[grep("^ERCC", rownames(filtered))]
condition1=args[2]
condition2=args[3]
x <- as.factor(rep(c(condition1, condition2), each=2))
print(x)
set <- newSeqExpressionSet(as.matrix(filtered), phenoData = data.frame(x, row.names=colnames(filtered)))
head(counts(set))
set1 <- RUVg(set, spikes, k=1)
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = counts(set1), colData = pData(set1), design = ~ W_1 + x) # use spike-in adjust
# dds <- DESeqDataSetFromMatrix(countData = counts(set), colData = pData(set), design = ~x) # use not adjust
dds <- DESeq(dds)
res <- results(dds)
res <- res[order(res$padj), ]
## Merge with normalized count data
resdata <- merge(as.data.frame(res), as.data.frame(counts(dds, normalized=TRUE)), by="row.names", sort=FALSE)
names(resdata)[1] <- "Transcript"
head(resdata)
resdata = resdata[resdata$pvalue>=0,]
## Write results
savefn=args[4]
write.csv(resdata, file=savefn)
加载.Rdata数据
> my = load('/Share2/home/zhangqf5/gongjing/rare_cell/tools/CellSIUS/Codes_Data/input/Supplementary_File_8_sce_raw.RData')
# 查看加载的变量
> my
[1] "sce"
# 不是list形式调用
> my$sce
Error in my$sce : $ operator is invalid for atomic vectors
# 通过ls命令查看环境定义的变量
# 核对一下刚加载的是哪个
> ls()
[1] "code_dir" "input_dir" "my" "out_data_dir" "plotdir"
[6] "sce"
# 查看加载的变量具有哪些属性可以使用
> attributes(sce)
$.__classVersion__
R Biobase eSet ExpressionSet SCESet
"3.4.1" "2.36.2" "1.3.0" "1.0.0" "1.1.9"
$logExprsOffset
[1] 1
$lowerDetectionLimit
[1] 0
$cellPairwiseDistances
dist(0)
$featurePairwiseDistances
dist(0)
$reducedDimension
<0 x 0 matrix>
$bootstraps
numeric(0)
$sc3
list()
$featureControlInfo
An object of class 'AnnotatedDataFrame'
rowNames: 1
varLabels: name
varMetadata: labelDescription
因为这个是biology的数据,所以有一些通用的属性,以及取值的方法:
# 使用exprs获取count数据
# 这也是我们一般使用的expression matrix数据
# 行:gene,列:cell id
> countdata<-exprs(sce)
> dim(countdata)
[1] 23848 11680
> head(countdata,n=2)
JRK_DA234_C2782 JRK_DA234_C2783 JRK_DA234_C2784 JRK_DA234_C2785
ENSG00000238009 0 0 0.9093836 0
ENSG00000239945 0 0 0.9093836 0
# fData: 获取feature data,这里是基因id
# 这个matrix相当于是对基因的注释,各种注释信息放进来
> fdata<-fData(sce)
> dim(fdata)
[1] 23848 15
> head(fdata, head=2)
gene_id chr symbol gene_biotype mean_exprs
ENSG00000238009 ENSG00000238009 1 RP11-34P13.7 lincRNA 0.0046305668
ENSG00000239945 ENSG00000239945 1 RP11-34P13.8 lincRNA 0.0004008153
ENSG00000279457 ENSG00000279457 1 FO538757.2 protein_coding 0.1828388937
ENSG00000228463 ENSG00000228463 1 AP006222.2 lincRNA 0.2726365525
ENSG00000236601 ENSG00000236601 1 RP4-669L17.2 lincRNA 0.0006401731
ENSG00000237094 ENSG00000237094 1 RP4-669L17.10 lincRNA 0.0098135864
exprs_rank n_cells_exprs total_feature_exprs pct_total_exprs
ENSG00000238009 7274 59 54.085020 7.842510e-05
ENSG00000239945 2200 6 4.681523 6.788366e-06
ENSG00000279457 16875 2019 2135.558279 3.096632e-03
ENSG00000228463 18491 2948 3184.394934 4.617480e-03
ENSG00000236601 3144 9 7.477221 1.084222e-05
ENSG00000237094 8776 120 114.622690 1.662068e-04
pct_dropout total_feature_counts log10_total_feature_counts
ENSG00000238009 99.49486 61 1.792392
ENSG00000239945 99.94863 6 0.845098
ENSG00000279457 82.71404 2307 3.363236
ENSG00000228463 74.76027 3724 3.571126
ENSG00000236601 99.92295 9 1.000000
ENSG00000237094 98.97260 121 2.086360
pct_total_counts is_feature_control_MT is_feature_control
ENSG00000238009 2.977997e-05 FALSE FALSE
ENSG00000239945 2.929177e-06 FALSE FALSE
ENSG00000279457 1.126269e-03 FALSE FALSE
ENSG00000228463 1.818042e-03 FALSE FALSE
ENSG00000236601 4.393765e-06 FALSE FALSE
ENSG00000237094 5.907174e-05 FALSE FALSE
> colnames(fdata)
[1] "gene_id" "chr"
[3] "symbol" "gene_biotype"
[5] "mean_exprs" "exprs_rank"
[7] "n_cells_exprs" "total_feature_exprs"
[9] "pct_total_exprs" "pct_dropout"
[11] "total_feature_counts" "log10_total_feature_counts"
[13] "pct_total_counts" "is_feature_control_MT"
[15] "is_feature_control"
# pData: 获取phenotype data,这里是cell id
# 这个matrix相当于是对细胞的注释,各种注释信息放进来
> pdata<-pData(sce)
> dim(pdata)
[1] 11680 38
> head(pdata, head=2)
cell_idx Batch cell_line cell_cycle_phase G1
IMR90_HCT116_C1 IMR90_HCT116_C1 IMR90_HCT116 HCT116 G1 1.000
IMR90_HCT116_C3 IMR90_HCT116_C3 IMR90_HCT116 HCT116 G1 0.972
IMR90_HCT116_C4 IMR90_HCT116_C4 IMR90_HCT116 IMR90 G1 0.999
> colnames(pdata)
[1] "cell_idx"
[2] "Batch"
[3] "cell_line"
[4] "cell_cycle_phase"
[5] "G1"
[6] "S"
[7] "G2M"
[8] "total_counts"
[9] "log10_total_counts"
[10] "filter_on_total_counts"
[11] "total_features"
[12] "log10_total_features"
[13] "filter_on_total_features"
[14] "pct_dropout"
[15] "exprs_feature_controls_MT"
[16] "pct_exprs_feature_controls_MT"
[17] "filter_on_pct_exprs_feature_controls_MT"
[18] "counts_feature_controls_MT"
[19] "pct_counts_feature_controls_MT"
[20] "filter_on_pct_counts_feature_controls_MT"
[21] "n_detected_feature_controls_MT"
[22] "n_detected_feature_controls"
[23] "counts_feature_controls"
[24] "pct_counts_feature_controls"
[25] "filter_on_pct_counts_feature_controls"
[26] "pct_counts_top_50_features"
[27] "pct_counts_top_100_features"
[28] "pct_counts_top_200_features"
[29] "pct_counts_top_500_features"
[30] "pct_counts_top_50_endogenous_features"
[31] "pct_counts_top_100_endogenous_features"
[32] "pct_counts_top_200_endogenous_features"
[33] "pct_counts_top_500_endogenous_features"
[34] "counts_endogenous_features"
[35] "log10_counts_feature_controls_MT"
[36] "log10_counts_feature_controls"
[37] "log10_counts_endogenous_features"
[38] "is_cell_control"
查看模块和环境版本
# 查看单个加载的模块的版本
> packageVersion('scater')
[1] ‘1.10.1’
# 查看整个运行环境:加载的包及版本
> sessionInfo()
R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Red Hat Enterprise Linux Server release 6.6 (Santiago)
Matrix products: default
BLAS: /Share/home/zhangqf5/software/R/3.5.1/lib64/R/lib/libRblas.so
LAPACK: /Share/home/zhangqf5/software/R/3.5.1/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.utf8 LC_NUMERIC=C
[3] LC_TIME=en_US.utf8 LC_COLLATE=en_US.utf8
[5] LC_MONETARY=en_US.utf8 LC_MESSAGES=en_US.utf8
[7] LC_PAPER=en_US.utf8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.utf8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] scater_1.10.1 ggplot2_3.1.0
[3] SingleCellExperiment_1.4.1 SummarizedExperiment_1.12.0
[5] DelayedArray_0.8.0 BiocParallel_1.16.6
[7] matrixStats_0.54.0 Biobase_2.28.0
[9] GenomicRanges_1.34.0 GenomeInfoDb_1.18.2
[11] IRanges_2.16.0 S4Vectors_0.20.1
[13] BiocGenerics_0.28.0
loaded via a namespace (and not attached):
[1] beeswarm_0.2.3 tidyselect_0.2.5 xfun_0.4
[4] reshape2_1.4.3 purrr_0.3.2 HDF5Array_1.10.1
[7] lattice_0.20-38 rhdf5_2.26.2 colorspace_1.4-0
[10] htmltools_0.3.6 viridisLite_0.3.0 yaml_2.2.0
[13] rlang_0.3.1 pillar_1.3.1 glue_1.3.0
[16] withr_2.1.2 GenomeInfoDbData_1.2.0 plyr_1.8.4
[19] stringr_1.3.1 zlibbioc_1.14.0 munsell_0.5.0
[22] gtable_0.2.0 evaluate_0.12 knitr_1.21
[25] vipor_0.4.5 Rcpp_1.0.1 scales_1.0.0
[28] backports_1.1.3 XVector_0.22.0 gridExtra_2.3
[31] digest_0.6.18 stringi_1.2.4 dplyr_0.8.0.1
[34] grid_3.5.1 rprojroot_1.3-2 tools_3.5.1
[37] bitops_1.0-6 magrittr_1.5 lazyeval_0.2.1
[40] RCurl_1.95-4.11 tibble_2.0.1 crayon_1.3.4
[43] pkgconfig_2.0.2 Matrix_1.2-16 DelayedMatrixStats_1.4.0
[46] ggbeeswarm_0.6.0 assertthat_0.2.0 rmarkdown_1.10
[49] viridis_0.5.1 Rhdf5lib_1.4.3 R6_2.3.0
[52] compiler_3.5.1
查看当前bioconductor的版本
有时候需要指定的bioconductor版本,才能正确的运行程序。因为不同的版本其安装的包的版本不同,如果不同版本差异很大,经常是容易报错的。比如最近遇到的这个:scRNAseq_workflow_benchmark,其强调说的bioconductor版本是3.5,如果是其他版本,跑出来报错,尤其是通过其安装的包scater,差异很大,已知运行不成功。
> tools:::.BioC_version_associated_with_R_version()
[1] ‘3.5’
从bioconductor安装指定版本的包
比如可能要安装低版本的包,需要先在bioconductor中找到对应的,然后指定安装url:
# 比如:安装低版本的scater
# 这里没有成功,因为还有几个其他依赖的包安装不成功
install_version('scater', version='1.4.0',repos = "https://bioconductor.org/packages/3.5/bioc")
从source文件安装包
# 先下载源文件,一般是压缩的
install.packages('/Share2/home/zhangqf5/gongjing/software/mvoutlier_2.0.9.tar.gz', repos = NULL, type="source")
移除安装包
# https://stat.ethz.ch/R-manual/R-devel/library/utils/html/remove.packages.html
remove.packages(pkgs, lib)
设置包的加载路径
在集群上,有的时候自己的目录下面安装了包,但是在系统调用的时候可能没有加载进来,可能是因为libpath中的路径存在问题,导致一些依赖包的顺序不对,所以不能正常的调用:
# 系统路径是在前的
# 直接调用时,依赖的包是系统的R安装的
# 与用户的R版本不一致,出现问题
> .libPaths()
[1] "/Share/home/zhangqf5/R/x86_64-pc-linux-gnu-library/3.3"
[2] "/Share/home/zhangqf/usr/R-3.3.0/lib64/R/library"
[3] "/Share/home/zhangqf/usr/R-3.3.2/lib64/R/library"
[4] "/Share/home/zhangqf5/software/R/3.5.1/lib64/R/library"
> library(mvoutlier)
Loading required package: sgeostat
Error: package or namespace load failed for ‘mvoutlier’:
package ‘rrcov’ was installed by an R version with different internals; it needs to be reinstalled for use with this R version
>
# 更换lib顺序,使得用户的libpath优先调用
> .libPaths("/Share/home/zhangqf5/software/R/3.5.1/lib64/R/library")
>
> library(mvoutlier)
sROC 0.1-2 loaded
>
移除已经导入的包
有时候会遇到这种问题,先导入了module1,module1默认是导入module2(v1)的,接下来要导入module3,module3是依赖于module2(v2)的,因为v2和v1版本不同,所以使得不能正常导入module2。此时可以先把module1 deattach掉,然后专门的导入module2:
比如下面先导入了模块m6ALogisticModel,其默认导入ggplot2,接下来导入caret,但是其依赖的ggplot2版本不同,所以导入时报错:
> library(caret)
Loading required package: ggplot2
Error in value[[3L]](cond) :
Package ‘ggplot2’ version 3.1.0 cannot be unloaded:
Error in unloadNamespace(package) : namespace ‘ggplot2’ is imported by ‘m6ALogisticModel’ so cannot be unloaded
可以先将m6ALogisticModel移除(参考这里),再导入:
> detach("package:m6ALogisticModel", unload=TRUE)
> library(caret)
Loading required package: ggplot2
df中取不含某列列名的其他列
参考这里:
# 提取training df中列名称不为class的其他列
Training.predictor.Gendata = training[,!names(training) %in% c("class")]
Read full-text »
Questions about ML
2018-05-25
参考
- 2017 41 Essential Machine Learning Interview Questions 中文版
- 2017 BAT机器学习面试1000题系列(第1~305题) @github
- 2018 Top 30 data science interview questions
- 2019 A Guide To Machine Learning Interview Questions And Answers
- 机器学习面试题总结@知乎
- Algorithm_Interview_Notes-Chinese@github
目录
- 参考
- 目录
- 算法
- 1. 什么是偏差(bias)、方差(variable)之间的均衡?
- 2. 监督学习和非监督学习有什么不同?
- 3. KNN和 k-means 聚类由什么不同
- 4. 定义精度和召回率
- 5. 解释一下ROC曲线的原理
- 6. 什么是贝叶斯定理?它在机器学习环境中如何有用?
- 7. 为什么我们要称“朴素”贝叶斯?
- 8. L1、L2正则化之间有什么不同?
- 9. 你最喜欢的算法是什么?把它解释一下。
- 10. 第一类误差和第二类误差有什么区别?
- 11. 什么是傅立叶变换?
- 12. 概率和似然有什么区别?
- 13. 什么是深度学习,它与其他机器学习算法相比怎么样?
- 14. 生成模型与判别模型有什么区别?
- 15. 交叉检验如何用在时间序列数据上?
- 16. 如何对决策树进行剪枝?
- 17. 模型的精度和模型的性能哪个对你更重要?
- 18. 什么是F1分数,怎么使用它?
- 19. 如何处理一个不平衡的数据集?
- 20. 什么时候你应该使用分类而不是回归?
- 21. 举个例子,说明使用集成学习会很有用
- 22. 你如何确保你的模型没有过拟合?
- 23. 如何评估你的机器学习模型的有效性?
- 24. 如何评估一个逻辑回归模型(LR model)?
- 25. 什么是核技巧,为什么它是有用的?
- 编程
- 持续兴趣
- 业务结合
算法
1. 什么是偏差(bias)、方差(variable)之间的均衡?
- 偏差是模型预测结合和真实结果之间的吻合程度,反应的是模型的准确性。方差是模型在不同的数据集上预测的效果差异大小,反应的是模型的鲁棒性。通常,如果模型属于高偏差、低方差的,那么模型的预测能力很弱,预测准确性差(偏差大),对于不同的数据集波动性不大(预测效果都很差,方差小),此时模型是欠拟合的。如果模型属于低偏差、高方差的,说明模型预测的效果很好,但是仅限于训练的数据集,当换其他的数据集时,可能效果不好,此时模型是过拟合的。所以在构建模型时,需要时刻关注模型的偏差、方差大小的变化,使得两者保持平衡。
- Bias 是由于你使用的学习算法过度简单地拟合结果或者错误地拟合结果导致的错误。它反映的是模型在样本上的输出与真实值之间的误差,即模型本身的精准度,即算法本身的拟合能力。Bias 可能会导致模型欠拟合,使其难以具有较高的预测准确性,也很难将你的知识从训练集推广到测试集。
- Variance 是由于你使用的学习算法过于复杂而产生的错误。它反映的是模型每一次输出结果与模型输出期望之间的误差,即模型的稳定性。反应预测的波动情况。Variance 过高会导致算法对训练数据的高纬度变化过于敏感,这样会导致模型过度拟合数据。从而你的模型会从训练集里带来太多噪音,这会对测试数据有一定的好处。
- Bias-Variance 的分解,本质上是通过在基础数据集中添加偏差、方差和一点由噪声引起的不可约误差,来分解算法上的学习误差。从本质上讲,如果你使模型更复杂并添加更多变量,你将会失去一些 Bias 但获得一些 Variance,这就是我们所说的权衡(tradeoff)。这也是为什么我们在建模的过程中,不希望这个模型同时拥有高的偏差和方差。
2. 监督学习和非监督学习有什么不同?
- 监督学习是根据有标签的数据集学习模型,去预测没有标签的数据集。非监督学习的数据集则是没有标签的,我们需要去学习数据本身潜在的结构。
- 监督学习需要train有label的数据。例如,为了进行classification(一项受监督的学习任务),您需要首先标记将用于培训模型的数据,以便将数据分类到标记的组中。相反的,无监督学习不需要明确标记数据。
3. KNN和 k-means 聚类由什么不同
- KNN是K最近邻分类,是属于监督学习的分类算法;k-mean是用于无标签数据的非监督的聚类算法。
- K-Nearest Neighbors是一种监督分类算法,而 k-means聚类是一种无监督的聚类算法。 虽然这些机制起初可能看起来相似,但这实际上意味着为了使K-Nearest Neighbors工作,你需要标记数据,以便将未标记的点分类(因此是最近邻居部分)。 K均值聚类仅需要一组未标记的点和阈值:算法将采用未标记的点并逐渐学习如何通过计算不同点之间的距离的平均值将它们聚类成组。
- 这里的关键区别在于,KNN需要标记点,因此是有监督的学习,而k-means不是,因此是无监督学习。
4. 定义精度和召回率
- 精度是precision,指在预测的结果中有多少的比例是预测对的,表达式是:TP/(TP+FN);召回率是recall,指样本集中的正样本,有多少被预测对了,表达式是:TP/(TP+FP)。
- 召回(率)也称为真阳性率:您的模型声称的阳性数量与整个数据中的实际阳性数量相比。精确度也称为阳性预测值,它衡量的是您的模型声称与实际声称的阳性数量相比的准确阳性数量。在您预测在10个苹果的情况下有10个苹果和5个橙子的情况下,可以更容易地想到召回率和精确度。你有完美的召回(实际上有10个苹果,你预测会有10个),但66.7%的精度,因为在你预测的15个事件中,只有10个(苹果)是正确的。
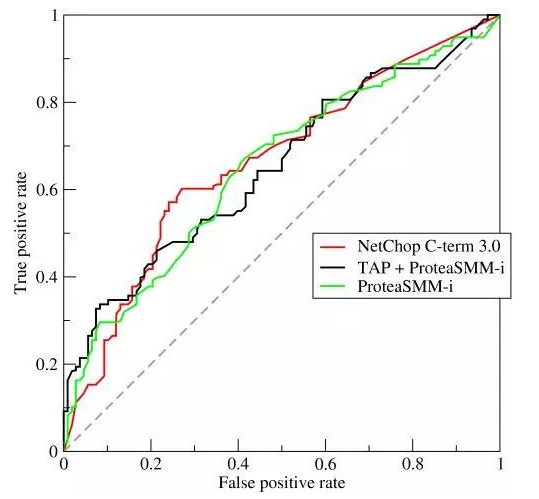
5. 解释一下ROC曲线的原理
- ROC是综合了模型评估的两个量值的曲线,一个是true positive rate,一个是false positive rate。曲线与坐标轴的围成的面积,可表示模型的整体效果,越接近于1则效果越好。
- ROC曲线是真阳率与各种阈值下的假阳率之间的对比度的图形表示。 它通常用作代表模型灵敏度(真阳性)与跌落之间的平衡或它将触发误报(假阳性)的概率。
6. 什么是贝叶斯定理?它在机器学习环境中如何有用?
- 贝叶斯定理是根据先验知识推测后验概率的模型,公式是:P(A|B)=P(B|A)P(A)/P(B)。
- 贝叶斯定理描述了当你不能准确知悉一个事物的本质时,你可以依靠与事物特定本质相关的事件出现的多少去判断其本质属性的概率。 它给出了已知先验知识下事件的后验概率。
- 在数学上,它表示为条件样本的真阳性率除以总体的假阳性率和条件的真阳性率之和。假设你在流感测试后有60%的机会真的感染了流感,但是在感染了流感的人中,50%的测试都是错误的,总人口只有5%的机会感染了流感。在做了阳性测试后,你真的有60%的机会患上流感吗?
- 贝叶斯定理说不,它说你有一个(0.6x0.05)(条件样本的真阳性率)/(0.6x0.05)(条件样本的真阳性率)+(0.5x0.95)(人群的假阳性率)= 5.94%的机会感染流感。
7. 为什么我们要称“朴素”贝叶斯?
- 因为其基本假设,是各变量是相互独立的,但是在现实情况中通常不是这样的,这种假设过于简单。
- 尽管 Naive Bayes 具有实际应用,特别是在文本挖掘中,但它被认为是“天真的”,因为它假设在实际数据中几乎不可能看到:条件概率被计算为组件个体概率的纯乘积。 这意味着特征的绝对独立性 – 这种情况在现实生活中可能永远不会遇到。正如 Quora 上一些评论者所说的那样,Naive Bayes 分类器发现你喜欢泡菜和冰淇淋之后,可能会天真地推荐你一个泡菜冰淇淋。
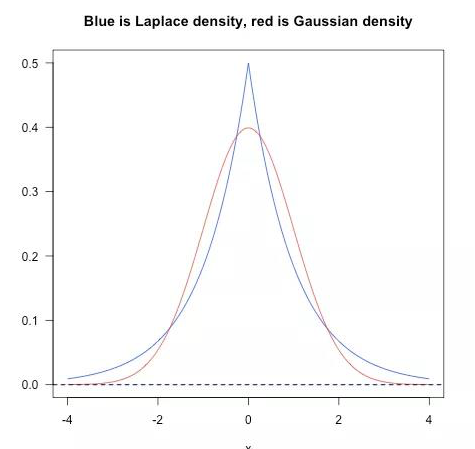
8. L1、L2正则化之间有什么不同?
- 对应不同的范式,L1是一级的,L2是二级的,对于权重的影响是更大的。
- L2正则,对应的是加入2范数,使得对权重进行衰减,从而达到惩罚损失函数的目的,防止模型过拟合。保留显著减小损失函数方向上的权重,而对于那些对函数值影响不大的权重使其衰减接近于0。相当于加入一个gaussian prior。
- L1正则 对应的是加入1范数,同样可以防止过拟合。它会产生更稀疏的解,即会使得部分权重变为0,达到特征选择的效果。相当于加入了一个laplacean prior。
9. 你最喜欢的算法是什么?把它解释一下。
- 最喜欢的是k-means聚类算法,因为这个在自己过去的项目中使用得比较多。比如我们有不同细胞发育时期的,每个基因的表达量数据,我们希望把基因根据表达谱的变化进行聚类,kmeans就是一个不错的选择。这个算法主要包含以下步骤:随机选取k个点作为起始的中心点,其中k是想要聚出的类的数目,然后对于其他所有的点,进行类分配,具体就是计算这个点到k个中心点的距离,距离最近的那个中心点所对应的类就是这个点的类别。然后更新中心点,就是对于每个类,重新计算中心点的值。接着再对其他的点重新进行类分配,不断迭代,直到类不发生变化未知。
- 这种类型的问题测试了你是否能优雅的,进行复杂的交流,或者技术上的细节,以及快速和有效地总结的能力。确保你有选择,确保你能简单有效地解释不同的算法,使一个五岁的孩子能够掌握基础知识!
10. 第一类误差和第二类误差有什么区别?
- x
- 第一类误差指的是假正率,第二类指的是假负率。简单来说,第一类误差意味着假设为真的情况下,作出了拒绝原假设的一种错误推断。第二类误差意味着假设为假的情况下,做出了接受原假设的一种错误判断。举个例子:第一类误差,你误判一个男的他怀孕了。第二类误差,你误判了一位其实已经怀孕的女子没怀孕。
11. 什么是傅立叶变换?
- x
- 傅立叶变换是将一般函数分解成对称函数叠加的一般方法。或者,正如这篇更直观的教程所说,在一杯冰沙中,我们就是这样找到配方的。傅立叶变换找到一组循环速度、振幅和相位,以匹配任何时间信号。傅立叶变换将信号从时间域转换为频率域-这是从音频信号或其他时间序列(如传感器数据)中提取特征的一种非常常见的方法。
12. 概率和似然有什么区别?
- 概率是某个事件出现的真实频率占所有可能发生情况的比例,似然则是对于某个时间的估计。
- 概率和似然都是指可能性,但在统计学中,概率和似然有截然不同的用法。概率描述了已知参数时的随机变量的输出结果;似然则用来描述已知随机变量输出结果时,未知参数的可能取值。例如,对于“一枚正反对称的硬币上抛十次”这种事件,我们可以问硬币落地时十次都是正面向上的“概率”是多少;而对于“一枚硬币上抛十次,我们则可以问,这枚硬币正反面对称的“似然”程度是多少。
- 概率(密度)表达给定θ下样本随机向量X=x的可能性,而似然表达了给定样本X=x下参数θ1(相对于另外的参数θ2)为真实值的可能性。我们总是对随机变量的取值谈概率,而在非贝叶斯统计的角度下,参数是一个实数而非随机变量,所以我们一般不谈一个参数的概率,而说似然。
13. 什么是深度学习,它与其他机器学习算法相比怎么样?
- 深度学习是机器学习的一个分支,依赖于神经网络进行建模,在大数据集上时有比较明显的优势,一般用于解决逻辑或者线性不可分的问题。
- 深度学习是与神经网络有关的机器学习的一个子集:如何使用反向传播和神经科学中的某些原理来更精确地建模大量未标记或半结构化数据。从这个意义上说,深度学习是一种无监督的学习算法,它通过使用神经网络来学习数据的表示。
14. 生成模型与判别模型有什么区别?
- x
- 生成模型将学习数据类别,而判别模型将简单地学习不同类别数据之间的区别。 判别模型通常优于分类任务的生成模型。
15. 交叉检验如何用在时间序列数据上?
- x
- 与标准的k-folds 交叉检验不同,数据不是随机分布的,而是具有时序性的。如果模式出现在后期,模型仍然需要选择先前时间的数据,尽管前期对模式无影响。我们可以如下这么做:
- fold1:training[1], test[2]
- fold2:training[1 2], test[3]
- fold3:training[1 2 3], test[4]
- fold4:training[1 2 3 4], test[5]
- fold5:training[1 2 3 4 5], test[6]
16. 如何对决策树进行剪枝?
- 可以采用自底向上的方法,依次合并两两最相似的节点,当树枝达到指定阈值,即可停止剪枝。
- 剪枝是在决策树中,为了降低模型的复杂度,提高决策树模型的预测精度,去除预测能力较弱的分支后所发生的现象。修剪可以自下而上和自上而下进行,方法包括减少错误修剪和成本复杂度修剪。
- 减少错误修剪可能是最简单的版本:替换每个节点。如果不降低预测精度,则保持修剪。虽然很简单,但这种启发式方法实际上非常接近于一种可以最大限度地优化准确性的方法。
17. 模型的精度和模型的性能哪个对你更重要?
- 这个得看是什么问题,如果是在线的学习模型,可能性能更重要,因为更看重反馈的及时性。如果是离线的模型,可能精度更加重要。
- 这个问题测试您对机器学习模型性能细微差别的理解!机器学习面试问题往往着眼于细节。有些模型具有更高的准确度,而在预测能力方面表现较差 — 这有什么意义?
- 好吧,这一切都与模型的准确性仅仅是模型性能的一个子集有关,在这一点上,有时是一个误导。例如,如果你想在一个拥有数百万样本的海量数据集中检测欺诈行为,那么一个更准确的模型很可能会预测,如果只有极少数的案例是欺诈行为,那么根本就不会有欺诈行为。然而,对于预测模型来说,这是无用的——一个旨在发现声称根本没有欺诈的欺诈的模型!这样的问题可以帮助您证明您理解模型的准确性并不是模型性能的全部。
18. 什么是F1分数,怎么使用它?
- F1=2xPxR/(P+R),其中P是precision,R是recall,是一个综合了准确率和召回率的指标,其可用于不均衡的数据集中,比单存的准确率或者召回率更健壮。
- F1分数是衡量模型性能的指标。它是模型精度和召回的加权平均值,结果趋向于1是最好的,结果趋向于0是最差的。你可以在分类测试中使用它,而真正的否定并不重要。
19. 如何处理一个不平衡的数据集?
- x
- 例如,当您有一个分类测试,并且90%的数据都在一个类中时,就会产生一个不平衡的数据集。这就导致了问题:如果您对其他类别的数据没有预测能力,那么90%的精度然而可能会出现偏差!下面是一些克服困难的策略(这里重要的是,您对不平衡数据集可能造成的损害以及如何平衡具有敏锐的感知。):
- 1.收集更多数据,甚至数据集中的不平衡。
- 2.对数据集重新取样以纠正不平衡。
- 3.在你的数据集中尝试一个不同的算法。
20. 什么时候你应该使用分类而不是回归?
- 当预测的标签是连续变量,使用回归模型;如果是离散的变量,则使用分类模型。
- 分类产生离散值并将数据集转换为严格的类别,而回归则提供连续的结果,使您能够更好地区分各个点之间的差异。如果您希望结果反映数据集中数据点对某些明确类别的归属性(例如:如果您希望知道某个名称是男性还是女性,而不仅仅是它们与男性和女性名称之间的关联性),则可以使用分类而不是回归。
21. 举个例子,说明使用集成学习会很有用
- 决策树模型对于某个分类问题效果不好,本身是属于一个弱的学习器。可以通过集成的方式,构建一个具有很多棵决策树的模型,即随机森林模型,从而提高模型的性能。这里的随机森林就属于决策树模型的一种集成。
- 集成学习通过组合一些基学习算法来优化得到更好的预测性能,通常可以防止模型的过拟合使模型更具有鲁棒性。你可以列举一些集成学习的例子,如bagging、boosting、stacking等,并且了解他们是如何增加模型预测能力的。
22. 你如何确保你的模型没有过拟合?
- 对拿到的数据集进行拆分,一部分训练集,一部分作为验证集,通过learning curve查看在两个数据集上的损失情况,确保没有发生过拟合。如果发现出现了过拟合,应该调整模型,比如可以添加正则化项。
- 过度拟合的训练数据以及数据携带的噪音,对于测试数据会带来不确定的推测。有如下三种方法避免过拟合:
- 1.保持模型尽可能地简单:通过考量较少的变量和参数来减少方差,达到数据中消除部分噪音的效果。
- 2.使用交叉检验的手段如:k-folds cross-validation。
- 3.使用正则化的技术如:LASSO方法来惩罚模型中可能导致过拟合的参数。
23. 如何评估你的机器学习模型的有效性?
- 把数据集进行拆分,分为训练集和验证集,用训练集训练模型,验证集验证模型的效果,为了避免数据集拆分的随机性,通过会进行多折拆分。
- 首先你需要将数据分成训练集和测试集,或者使用给交叉验证方法分割。然后你需要选择度量模型表现的metrics,如F1数、准确率、混淆矩阵等。更重要的是,根据实际情况你需要理解模型度量的轻微差别,以便于选择正确的度量标准。
24. 如何评估一个逻辑回归模型(LR model)?
- x
- 上述问题的一部分。你必须演示对逻辑回归的典型目标(分类、预测等)的理解,并提供一些示例和用例。
25. 什么是核技巧,为什么它是有用的?
- 核技巧是指在模型的中间引入不同的模型,一般是非线性的,使得整个模型可以解决线性不可分问题。
- 核技巧使用核函数,确保在高维空间不需要明确计算点的坐标,而是计算数据的特征空间中的内积。这使其具有一个很有用的属性:更容易的计算高维空间中点的坐标。许多算法都可以表示称这样的内积形式,使用核技巧可以保证低维数据在高维空间中运用算法进行计算。
编程
1. 如何处理数据集中丢失或损坏的数据?
- 可以进行缺失值填补(imputation),常见的做法是平均值或者中位值填充,如果是离散类型的,可用频率最高的类别填充。更加复杂的,可以用其他的数据进行拟合,得到和其他特征之间的关系模型,然后使用关系模型对缺失值进行填充。
- **
2. 你是否有使用Spark或大数据工具进行机器学习的经验?
- 目前还没有使用过这些工具,因为之前处理的样本数量还不够大。
- **
3. 选择一个算法,实现一个并行的伪代码。
- x
- **
4. 链表和数组之间有什么区别?
- 链表是收尾相连的,但是可以有环形,数组就只是按顺序存储的系列元素。
- **
5. 描述哈希表
- 哈希表就是通过一个函数,将值映射到储存中的一些单元,方便快速的取值。
- **
6. 你使用哪些数据可视化库? 你对最佳数据可视化工具有何看法?
- 我现在主要使用python语言,里面有一些很强大的可视化模块,我主要使用的是seaborn和matplotlib。我觉得可视化的工具能给人更直观的感受,所谓一图值千言。最佳的可视化工具,我觉得应该是交互式的实时工具,方便看的人也能进行探索,有一种身临其境的感受。
- **
持续兴趣
1. 你读过的最近的一篇机器学习论文是什么?
- 看的是应用类型的,不是算法类型的。
- 如果你想表现出对机器学习职位的兴趣,就必须掌握最新的机器学习科学文献。这篇深入学习的后代(从Hinton到Bengio再到LeCun)对自然的深入学习的概述可以是一篇很好的参考论文,也可以是一篇深入学习中正在发生的事情的概述,以及你可能想引用的那种论文。
2. 你在机器学习方面有研究经验吗?
- x
- 与最后一点相关的是,大多数为机器学习职位招聘的组织都会寻找你在该领域的正式经验。由该领域的先行者共同撰写或监督的研究论文,可以使你在被雇佣和不被雇佣之间产生差异。确保你已经准备好了一份关于你的研究经验和论文的总结,如果你不准备的话,还要对你的背景和缺乏正式研究经验做出解释。
3. 你最喜欢的机器学习模型的案例是什么?
- 智能诊断系统。,链接:登上《Cell》封面的AI医疗影像诊断系统:机器之心专访UCSD张康教授
- 这里我们拿 Quora 上面的一个帖子为例,帖子在这里。上面的 Quora 帖子里包含一些示例,例如决策树,它根据智商分数将人们分类为不同的智力层次。确保你心里有几个例子,并描述与你产生共鸣的地方。重要的是你要对机器学习的实现方式表现出兴趣。
4. 你想以什么方式赢得“Netflix奖”比赛?
- x
- Netflix奖是一项著名的竞赛,Netflix提供了 $1,000,000的奖金,以获得更好的协同过滤算法(collaborative filtering algorithm)。关于这个比赛的最后赢家, BellKor;他们让这个算法效率提升百分之十,并且给出了多种解法。多了解这些行业相关的 Case 并且和你面试官侃侃而谈能够体现你对于机器学习这个领域的关注
5. 您通常在哪里寻找数据集?
- x
- 像这样的机器学习面试问题试图让你了解机器学习兴趣的核心。 真正热衷于机器学习的人将会独自完成侧面项目,并且很清楚那些伟大的数据集是什么。 如果您遗失任何内容,请查看 Quandl 获取的经济和财务数据,以及 Kaggle 的数据集集合,以获取其他优秀列表。
6. 你认为谷歌是如何为自动驾驶汽车提供培训数据的?
- x
- 像这样的机器学习面试问题确实测试了你对不同机器学习方法的知识,如果你不知道答案,你的创造力。谷歌目前正在使用 recaptcha 来获取店面和交通标志上的标签数据。他们还建立在由Sebastian Thrun在谷歌(Googlex)收集的培训数据的基础上 — 其中一些数据是由他在沙漠沙丘上驾驶马车的研究生获得的!
7. 你将如何模拟阿尔法戈在围棋中击败李世乭的方法?
- 通过蒙特卡洛采样,进行博弈,属于增强化学习的一种。
- 在五个系列赛中,阿尔法戈击败了围棋中最优秀的人类选手李思多,这是机器学习和深度学习史上一个真正具有开创性的事件。上面的 Nature 论文描述了这是如何通过“蒙特卡洛树搜索(Monte Carlo Tree Search)和深神经网络(Deep Neural Networks)来实现的,这些神经网络经过有监督的学习、人类专家游戏和加强自玩游戏的学习。”
业务结合
1. 您如何为我们公司的用户实施推荐系统?
- x
- 许多这种类型的机器学习面试问题将涉及机器学习模型的实施以解决公司的问题。 您必须深入研究公司及其行业,尤其是公司的收入驱动因素,以及公司在其所在行业中所采用的用户类型。
2. 我们如何利用您的机器学习技能来创造收入?
- x
- 这是一个棘手的问题。理想的答案将证明您对推动业务发展的因素以及您的技能如何关联的了解。例如,如果你正在面试音乐流初创公司Spotify,你可以说,你在开发更好的推荐模式方面的技能将增加用户保留率,从长远来看这将增加收入。上面链接的Startup Metrics Slideshare将帮助您准确了解在考虑支出和成长时,哪些绩效指标对初创技术公司是重要的。
3. 你认为我们当前的数据处理过程如何?
- x
- 这类问题要求你认真倾听,并以富有建设性和洞察力的方式传达反馈。 你的面试官正在试图判断您是否是他们团队中的重要成员,以及你是否根据公司或行业特定条件,掌握了为什么某些事情按照公司数据流程的方式设置的细微差别。 他们试图看看你是否可以成为有见地同行。 随行而动。这一系列的机器学习面试问题试图衡量你对机器学习的热情和兴趣。正确的答案将作为你承诺终身学习机器学习的证明。
Read full-text »
Google ML rules
2018-05-22
目录
机器学习规则(google: Rules of ML):
术语
- 实例:要对其进行预测的事物。
- 标签:预测任务的答案,训练数据的或者对新样本预测的结果。
- 特征:使用的实例的属性。
- 特征列:一组相关特征。
- 样本:一个实例和一个标签。
- 模型:预测任务的统计表示法。
- 指标:关心的某个数值。比如评估模型好坏的某些量值。
- 目标:算法尝试优化的一种指标。
- 管道:pipeline,机器学习算法的基础架构。
- 点击率:点击广告中的链接的网页访问者所占的百分比。
概览
进行机器学习的基本方法是:
- 确保管道从头到尾都稳固可靠。
- 从制定合理的目标开始。
- 以简单的方式添加常识性特征。
- 确保管道始终稳固可靠。
机器学习之前
- 【1】产品并非需要机器学习技术,在未获得足够数据前,请勿使用。
- 【2】首先设计并实现指标。
- 【3】选择机器学习技术而非复杂的启发式算法。
第一阶段:第一个pipeline
- 【4】自己搭建的第一个模型应简单易用。
- 【5】模型部分单独封装,其他的数据部分单独测试。
- 【6】复制pipeline时注意丢弃的数据。在更新机器学习算法时注意,旧数据的可用性取舍。
- 【7】启发式算法的信息转为特征。1)启发式算法预处理,比如发件人已加入黑名单,不要再学习黑名单的定义。2)创建特征。3)挖掘启发式算法的原始输入。4)修改标签。比如根据常识进行适当的变化等。
监控
- 【8】系统对于实时更新程度的要求,不同的产品对于更新的要求不同。比如微博等需要实时更新。
- 【9】导出模型用于新数据的预测之前需要先进行检测评估。
- 【10】注意隐藏的问题。比如数据的实时更新,可能提升模型的效果。
- 【11】记录特征的来源信息(作者、文档等)。
第一个目标
- 【12】选择优化目标时,不要纠结。
- 【13】可观察可归因的简单指标。
- 【14】选择可解释的模型,方便调试。
- 【15】在策略层区分垃圾内容和质量排名。
特征工程
- 【16】制定发布和迭代模型计划。提前规划添加新特征、调整目标等。
- 【17】从可直接观察和报告的特征着手。
- 【18】探索可跨情景泛化的内容的特征。
- 【19】使用具体的特征。
- 【20】创建新特征时尽可能简单。
- 【21】在线性模型中学习的特征权重数目与数据量大致成正比。
- 【22】清理不使用的特征。
人工分析
- 【23】让用户参与评估,以体验的方式。
- 【24】衡量模型间的差异。
- 【25】实用比预测效果更重要。
- 【26】基于效果评估寻找规律,创建新特征以优化模型。
- 【27】尝试量化观察到的异常行为,先量化再优化。
- 【28】关注数据行为的稳定性或者模型的稳定性。
训练-应用偏差
- 【29】应用时的一些特征可以记录下来,在训练时使用,防止训练模型效果很好,但是应用效果很差。
- 【30】按重要性加权对数据采样,不能随意丢弃数据。
- 【31】注意训练或者应用期间数据是否变化。
- 【32】训练和应用时使用相同的代码。
- 【33】用当前的数据训练模型,明天及之后的模型进行测试。
- 【34】二元分类,可在短期内牺牲一点效果,以获得更高质量的数据。
- 【35】排名问题中存在的股友偏差。
- 【36】通过位置特征避免出现反馈环。
- 【37】训练-应用数据之间的偏差。
缓慢增长、优化细化和复杂模型
- 【38】如果目标不协调,并成为问题,就不要在新特征上浪费时间。
- 【39】发布决策代表的是长期产品目标。
- 【40】保证集成学习简单化。
- 【41】效果达到平稳后,寻找与现有信号有质的差别的新信息源并添加进来,而不是优化现有信号。
- 【42】不要期望多样性、个性化或相关性与热门程度之间的联系有您认为的那样密切。
- 【43】在不同的产品中,您的好友基本保持不变,但您的兴趣并非如此。
Read full-text »
Using DESeq2 to do differential expression analysis
2018-05-21
DESeq2 (full manual)
Calling differential expression (DE) genes based on read count among different conditions, for instance, medicine treatment versus mock. Generally we used both fold change and pvalue to filter DE genes, e.g., |log2(Fold change)| >=2 and pvalue <=0.05. Here is an simple script to run DE analysis as described here: Template for analysis with DESeq2.
Prepare read count data
[zhangqf7@loginview02 RIP]$ head readcount_h2.txt
NM_212847 1922.0 3057.0
NM_212844 1552.0 245.0
NM_001004627 1901.0 22507.0
NM_212849 15338.0 2889.0
NM_205638 3326.0 13646.0
Load read count and condition data, run the DE analysis
library("DESeq2")
countdata <- read.table("/Share/home/zhangqf7/gongjing/zebrafish/data/RIP/readcount_h2.txt", header=FALSE, row.names=1)
colnames(countdata) = c("HuR", "Control")
countdata <- as.matrix(countdata)
condition = factor(c("HuR", "Control"))
(coldata <- data.frame(row.names=colnames(countdata), condition))
dds <- DESeqDataSetFromMatrix(countData=countdata, colData=coldata, design=~condition)
dds <- DESeq(dds)
# Get differential expression results
res <- results(dds)
#table(res$padj<0.05)
## Order by adjusted p-value
res <- res[order(res$padj), ]
## Merge with normalized count data
resdata <- merge(as.data.frame(res), as.data.frame(counts(dds, normalized=TRUE)), by="row.names", sort=FALSE)
names(resdata)[1] <- "Transcript"
head(resdata)
## filter by pvalue or any criteria else
resdata = resdata[resdata$pvalue<0.05,]
## Write results
write.csv(resdata, file="/Share/home/zhangqf7/gongjing/zebrafish/data/RIP/readcount_h2.DE.csv")
Finally the content of result object resdata, and can be saved to a csv file:
Transcript baseMean log2FoldChange lfcSE stat pvalue
1 NM_212844 1139.1833 2.539801 1.121616 2.264413 0.02354874
2 NM_001004627 9612.8973 -2.147552 1.010351 -2.125550 0.03354078
3 NM_212849 11431.3248 2.497761 1.064955 2.345415 0.01900593
4 NM_001159988 1066.9555 -2.126481 1.068342 -1.990450 0.04654139
5 NM_001159983 1337.8418 -2.272092 1.083839 -2.096337 0.03605233
6 ENSDART00000172192 307.6127 -2.232198 1.097730 -2.033467 0.04200536
padj HuR Control
1 0.3812277 2097.04486 181.3218
2 0.3812277 2568.60972 16657.1849
3 0.3812277 20724.53227 2138.1173
4 0.3812277 268.88655 1865.0245
5 0.3812277 291.85676 2383.8269
6 0.3812277 67.55943 547.6659
Read full-text »
Circos tableview tool to visualize interaction correlation or distribution
2018-05-19
Circos tableview
Circos can show the connections between different elements, e.g., genomic regions, RNA molecules and so on. Another userful tool tableviewer can be used to display the percentage of each crosslinks among multiple relationships. In RISE database we collected RNA-RNA interactions (RRIs) from various sources, and tableviewer are used for visualization of the landscape of RRIs. Here is the example:

Generate stats data from data frame
Generally we can read any txt file into pandas data frame, and use function groupby & count to count the entry for each pair of two variables. Here is the saved data frame:
[zhangqf5@loginview02 data]$ cat RRI_union_deduplicates.split_full.type_dis.human.txt
data proteinCoding lncRNA miRNA rRNA snoRNA snRNA tRNA NoncanonicalRNA others
proteinCoding 41863.0 4756.0 183.0 14128.0 115.0 721.0 66.0 3520.0 2762.0
lncRNA 9982.0 1080.0 1984.0 194.0 40.0 91.0 2.0 748.0 482.0
miRNA 35798.0 304.0 36.0 14.0 6.0 23.0 1.0 458.0 117.0
rRNA 1347.0 147.0 3.0 360.0 31.0 15.0 27.0 100.0 83.0
snoRNA 105.0 21.0 2.0 290.0 299.0 86.0 6.0 24.0 10.0
snRNA 969.0 170.0 1.0 41.0 29.0 851.0 1.0 70.0 52.0
tRNA 22.0 1.0 0.0 3.0 1.0 1.0 210.0 3.0 23.0
NoncanonicalRNA 6010.0 705.0 1076.0 69.0 104.0 69.0 1.0 1132.0 271.0
others 2850.0 384.0 328.0 74.0 12.0 43.0 5.0 262.0 400.0
Define color for your data
Then we can add header (more details can be found here) to define color for each element (RNA type here):
data 1 2 3 4 5 6 7 8 9
data 202,75,78 83,169,102 205,185,111 98,180,208 129,112,182 238,130,238 255,140,0 74,113,178 169,169,169
Concatenate data for plot
Once concatenate header and data frame, the combined data is ready for plot:
[zhangqf5@loginview02 data]$ cat RRI_union_deduplicates.split_full.type_dis.human.txt
data 1 2 3 4 5 6 7 8 9
data 202,75,78 83,169,102 205,185,111 98,180,208 129,112,182 238,130,238 255,140,0 74,113,178 169,169,169
data proteinCoding lncRNA miRNA rRNA snoRNA snRNA tRNA NoncanonicalRNA others
proteinCoding 41863.0 4756.0 183.0 14128.0 115.0 721.0 66.0 3520.0 2762.0
lncRNA 9982.0 1080.0 1984.0 194.0 40.0 91.0 2.0 748.0 482.0
miRNA 35798.0 304.0 36.0 14.0 6.0 23.0 1.0 458.0 117.0
rRNA 1347.0 147.0 3.0 360.0 31.0 15.0 27.0 100.0 83.0
snoRNA 105.0 21.0 2.0 290.0 299.0 86.0 6.0 24.0 10.0
snRNA 969.0 170.0 1.0 41.0 29.0 851.0 1.0 70.0 52.0
tRNA 22.0 1.0 0.0 3.0 1.0 1.0 210.0 3.0 23.0
NoncanonicalRNA 6010.0 705.0 1076.0 69.0 104.0 69.0 1.0 1132.0 271.0
others 2850.0 384.0 328.0 74.0 12.0 43.0 5.0 262.0 400.0
Calling tableviewer to plot
I wrote a python script to call tableviewer:
import subprocess, os
import sys
def tableview(txt=None, parse_table=None, make_conf=None, conf_dir=None, circos_conf=None, save_dir=None, parsed_conf=None):
if parse_table is None:
parse_table = '/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer/bin/parse-table'
if make_conf is None:
make_conf = '/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer/bin/make-conf'
if conf_dir is None:
conf_dir = '/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer2/data'
if circos_conf is None:
circos_conf = '/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer2/etc/circos.conf'
if save_dir is None:
save_dir = os.path.dirname(txt)
if parsed_conf is None:
#parsed_conf = '/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer/samples/parse-table-02a.conf'
parsed_conf = '/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer2/etc/parse-table.conf'
print "[tableview start] file: %s"%(txt)
subprocess.call(["cat {txt} | {parse_table} -conf {parsed_conf} -segment_order=ascii,size_desc -placement_order=row,col -interpolate_type count -col_order_row -use_col_order_row -col_color_row -use_col_color_row -ribbon_layer_order=size_asc | {make_conf} -dir {conf_dir}".format(txt=txt, parse_table=parse_table, parsed_conf=parsed_conf, make_conf=make_conf, conf_dir=conf_dir)],shell=True)
#subprocess.call(["cat {txt} | {parse_table} -conf {parsed_conf} | {make_conf} -dir {conf_dir}".format(txt=txt, parse_table=parse_table, parsed_conf=parsed_conf, make_conf=make_conf, conf_dir=conf_dir)],shell=True)
file_png = txt.split('/')[-1].replace('txt', 'png')
subprocess.call(["circos -conf {circos_conf} -outputdir {save_dir} -outputfile {file_png} -param random_string=zgvickusamp| grep created".format(circos_conf=circos_conf, save_dir=save_dir, file_png=file_png)],shell=True)
print "[tableview end] file: %s"%(txt)
def main():
#tableview(txt='/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer/samples/RRI_union_deduplicates.split_full.type_dis.txt')
if len(sys.argv) == 1:
txt = '/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer2/samples/RRI_union_deduplicates.split_full.type_dis.txt'
else:
txt = sys.argv[1]
tableview(txt)
if __name__ == '__main__':
main()
Run the script with previous data:
[zhangqf5@loginview02 tableviewer2]$ pwd
/Share/home/zhangqf5/gongjing/software/circos-tools-0.22/tools/tableviewer2
[zhangqf5@loginview02 tableviewer2]$ ll
total 40K
drwxr-x--- 2 zhangqf5 zhangqf 4.0K Jul 6 2017 batch
drwxr-x--- 2 zhangqf5 zhangqf 4.0K Aug 2 2017 bin
drwxr-x--- 2 zhangqf5 zhangqf 4.0K May 19 02:21 data
drwxr-x--- 2 zhangqf5 zhangqf 4.0K May 19 02:21 etc
drwxr-x--- 2 zhangqf5 zhangqf 4.0K Jul 6 2017 img
drwxr-x--- 2 zhangqf5 zhangqf 4.0K Jul 6 2017 lib
-rwxr----- 1 zhangqf5 zhangqf 2.4K Jul 6 2017 makeimage.py
drwxr-x--- 2 zhangqf5 zhangqf 4.0K Jul 6 2017 results
drwxr-x--- 2 zhangqf5 zhangqf 4.0K Jul 6 2017 samples
drwxr-x--- 2 zhangqf5 zhangqf 4.0K Jul 6 2017 uploads
[zhangqf5@loginview02 tableviewer2]$ python makeimage.py /Share/home/zhangqf5/gongjing/DNA-RNA-Protein-interaction-correlation-12-18/data/RRI_union_deduplicates.split_full.type_dis.human.txt
We get plot like this:

Issue: link direction ?
However, there is a issue. The connection between RNAs actually has no direction, thus the ribbon color from RNA1 to RNA2 must be the same as from RNA2 to RNA1. In the graph above, for example, there are two bands connect mRNA (red) and others (grey), but these two colors are different. In this case, we need to parse the data table to make all count values apear in only one side of the diagonal (upper or lower).
Data format conversion
Here is the function to transform the data format:
from nested_dict import nested_dict
def read_txt(txt='/Share/home/zhangqf5/gongjing/DNA-RNA-Protein-interaction-correlation-12-18/data/type_dis/RRI_union_deduplicates.split_full.type_dis.human.txt'):
dis_dict = nested_dict(2, int)
dis_revise_dict = nested_dict(2, int)
with open(txt, 'r') as TXT:
for n,line in enumerate(TXT):
line = line.strip()
print n,line
if n == 0:
dis_dict['row_order'] = line
elif n == 1:
dis_dict['row_color'] = line
elif n == 2:
dis_dict['row_rnas'] = line
col_rna_ls = line.split()[1:]
else:
row_rna = line.split()[0]
val_ls = map(float, line.split()[1:])
for col_rna, val in zip(col_rna_ls, val_ls):
dis_dict[row_rna][col_rna] = val
print dis_dict
rna_pair_ls = []
for row_rna in col_rna_ls:
for col_rna in col_rna_ls:
dis_revise_dict[row_rna][col_rna] = 0
for row_rna in col_rna_ls:
for col_rna in col_rna_ls:
rna_pair1 = row_rna + '-' + col_rna
rna_pair2 = col_rna + '-' + row_rna
if rna_pair1 in rna_pair_ls:
continue
if rna_pair2 in rna_pair_ls:
continue
rna_pair_ls.append(rna_pair1)
rna_pair_ls.append(rna_pair2)
dis_revise_dict[row_rna][col_rna] += dis_dict[row_rna][col_rna]
if row_rna == col_rna:
continue
dis_revise_dict[row_rna][col_rna] += dis_dict[col_rna][row_rna]
print dis_revise_dict
savefn = txt.replace('txt', 'revise.txt')
with open(savefn, 'w') as SAVEFN:
print >>SAVEFN, dis_dict['row_order']
print >>SAVEFN, dis_dict['row_color']
print >>SAVEFN, dis_dict['row_rnas']
for row_rna in col_rna_ls:
row_rna_ls = [dis_revise_dict[row_rna][col_rna] for col_rna in col_rna_ls]
print >>SAVEFN,row_rna+' '+' '.join(map(str, row_rna_ls))
return savefn
After converting, we get the data with all value on upper triangle:
[zhangqf5@loginview02 data]$ cat RRI_union_deduplicates.split_full.type_dis.human.revise.txt
data 1 2 3 4 5 6 7 8 9
data 202,75,78 83,169,102 205,185,111 98,180,208 129,112,182 238,130,238 255,140,0 74,113,178 169,169,169
data proteinCoding lncRNA miRNA rRNA snoRNA snRNA tRNA NoncanonicalRNA others
proteinCoding 41863.0 14738.0 35981.0 15475.0 220.0 1690.0 88.0 9530.0 5612.0
lncRNA 0 1080.0 2288.0 341.0 61.0 261.0 3.0 1453.0 866.0
miRNA 0 0 36.0 17.0 8.0 24.0 1.0 1534.0 445.0
rRNA 0 0 0 360.0 321.0 56.0 30.0 169.0 157.0
snoRNA 0 0 0 0 299.0 115.0 7.0 128.0 22.0
snRNA 0 0 0 0 0 851.0 2.0 139.0 95.0
tRNA 0 0 0 0 0 0 210.0 4.0 28.0
NoncanonicalRNA 0 0 0 0 0 0 0 1132.0 533.0
others 0 0 0 0 0 0 0 0 400.0
Comparison of original & transformed data frame
That is:
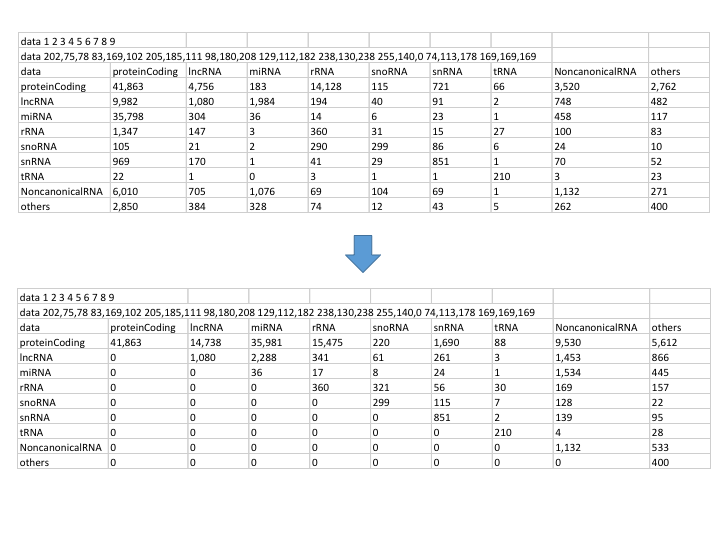
Then using these data we can visualize the links without direction as below:
Read full-text »
Latest articles
Links
- ZhangLab , RISE database , THU life , THU info
- Data analysis: pandas , numpy , scipy
- ML/DL: sklearn , sklearn(中文) , pytorch
- Visualization: seaborn , matplotlib , gallery
- Github: me